Généralité
Le terme rétinite pigmentaire (RP) identifie un groupe de maladies génétiques caractérisées par une dégénérescence rétinienne progressive.

La rétinite pigmentaire est une dystrophie rétinienne caractérisée par la perte progressive des photorécepteurs et un dysfonctionnement de l'épithélium pigmentaire, ce qui signifie que la rétine réduit progressivement sa capacité à transmettre des informations visuelles au cerveau via le nerf optique.
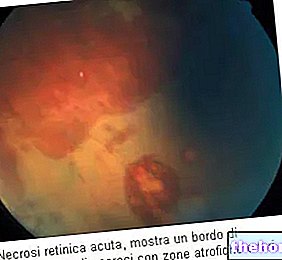
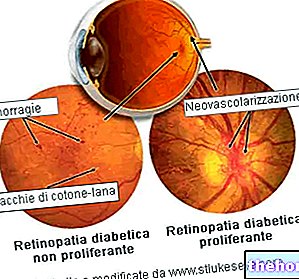
Le processus pathologique commence par des altérations de l'épithélium pigmentaire rétinien. Au fur et à mesure que la rétinite pigmentaire progresse, il se produit un amincissement des vaisseaux sanguins qui alimentent la rétine, qui s'atrophient. À l'examen du fond d'œil, les dépôts caractéristiques sont visuellement détectables. pigment rétinien ( d'où le nom de la maladie). Des modifications et des lésions atrophiques peuvent également impliquer le nerf optique et, progressivement, les cellules photosensibles de la rétine meurent.
Les patients atteints de rétinite pigmentaire éprouvent initialement des problèmes de vision surtout dans des environnements mal éclairés et se plaignent d'un rétrécissement du champ visuel périphérique. La vision centrale est épargnée jusqu'aux derniers stades de la maladie, et le résultat final peut varier considérablement : de nombreuses personnes atteintes de rétinite pigmentaire conservent une vision limitée tout au long de leur vie, tandis que d'autres perdent complètement la vue.
La rétinite pigmentaire est une maladie héréditaire, principalement causée par des modifications génétiques transmises par l'un ou les deux parents. Le type d'anomalie génétique détermine quelles cellules rétiniennes sont les plus impliquées dans la maladie et permet de distinguer, d'un point de vue clinique, les différentes affections. À ce jour, plus de 50 défauts génétiques différents impliqués dans la rétinite pigmentaire ont été identifiés. Les anomalies peuvent être transmises des parents à la progéniture par l'un des trois modes de transmission : autosomique récessif, autosomique dominant ou hétérosomal récessif (lié à l'X ou lié à l'X).
Symptômes
Pour plus d'informations : Symptômes de la rétinite pigmentaire
La rétinite pigmentaire survient généralement chez les adolescents et les jeunes adultes. Les symptômes apparaissent souvent entre 10 et 30 ans, mais le diagnostic peut être posé dès la petite enfance ou bien plus tard dans la vie.
Les premiers symptômes de la rétinite pigmentaire peuvent inclure :
- Difficulté à voir la nuit (cécité nocturne) ou dans des conditions de faible luminosité
- Adaptation lente de la vision dans l'obscurité à celle dans la lumière, et vice versa ;
- Rétrécissement du champ visuel et perte de la vision périphérique ;
- Sensibilité à la lumière et à l'éblouissement.
Certains symptômes dépendent du type de photorécepteurs impliqués. Les bâtonnets sont responsables de la vision en noir et blanc, tandis que les cônes permettent de distinguer les couleurs.
Dans la plupart des cas de rétinite pigmentaire, les bâtonnets sont impliqués en premier. Cependant, dans les formes à évolution rapide, les cônes peuvent également être affectés à un stade précoce.
Les bâtonnets sont concentrés dans les parties externes de la rétine et sont activés par une faible lumière, de sorte que leur dégénérescence affecte la vision périphérique et nocturne. Si des cônes sont impliqués, il est possible de ressentir une perte de perception des couleurs et de vision centrale.
La prédominance des photorécepteurs impliqués est déterminée par le défaut particulier présent dans la constitution génétique du patient.
Souvent, le premier symptôme de la rétinite pigmentaire est la cécité nocturne (ou noctalopie). Certaines personnes trouvent qu'elles ont besoin de plus en plus de temps pour s'adapter aux différences de luminosité lorsqu'elles passent d'une zone bien éclairée à une zone plus sombre. Une forme typique de perte de vision induit un rétrécissement de la vision périphérique (vision en tunnel ou en télescope) ; ce modèle est appelé un scotome en anneau. Parfois, ce phénomène peut manquer dans les premiers stades, mais il est remarqué lorsque l'individu trébuche souvent sur des objets ou est impliqué dans un accident de la circulation.Lorsque la perte de vision concerne la zone centrale de la rétine (également appelée dystrophie maculaire) les patients éprouvent des difficultés à lire et à effectuer des travaux détaillés qui nécessitent une concentration sur un seul objet, comme passer un fil dans le chas d'une aiguille.
Le taux de progression de la maladie et le degré de perte visuelle varient d'une personne à l'autre. Certains cas extrêmes peuvent évoluer rapidement en deux décennies, d'autres une évolution lente qui ne conduit jamais à la cécité complète. L'apparition précoce est observée dans les formes plus graves de rétinite pigmentaire, tandis que les patients atteints d'affections plus bénignes (p. ex., autosomique dominante) peuvent développer la maladie au cours de leur cinquième ou sixième décennie de vie. que les femmes et plus sévèrement; les femmes, en revanche, transmettent la caractéristique génétique (elles portent le gène altéré sur le chromosome X) et manifestent moins fréquemment les symptômes de la maladie.
Complications
La rétinite pigmentaire continuera à progresser, quoique lentement. Cependant, la cécité complète est rare, mais une réduction significative de la vision périphérique et centrale peut survenir.
Les patients atteints de rétinite pigmentaire développent souvent un gonflement de la rétine (œdème maculaire) ou des cataractes à un âge précoce. Ces complications peuvent être traitées si elles interfèrent avec la vision.
Maladies associées
Communément, un patient atteint de rétinite pigmentaire n'a pas d'autres troubles et dans ce cas on parle de rétinite pigmentaire « non syndromique » ou simple. Cependant, plusieurs syndromes partagent certains symptômes cliniques avec cette maladie oculaire ; le plus courant est le syndrome d'Usher, qui affecte environ 10 à 30 % de tous les patients atteints de rétinite pigmentaire et est associé à une perte auditive congénitale ou progressive concomitante. Dans l'amaurose congénitale de Leber, cependant, les enfants peuvent devenir aveugles, ou presque aveugles, au cours des six premiers mois de la vie.D'autres maladies liées à la rétinite pigmentaire comprennent le syndrome de Bardet-Biedl et la maladie de Refsum.
Causes
La maladie peut être causée par un certain nombre de défauts génétiques : en effet, il existe plusieurs gènes qui, s'ils sont affectés par l'altération, peuvent provoquer le phénotype de la rétinite pigmentaire.Ceux-ci codent normalement pour des protéines impliquées dans la cascade de transduction qui permet la vision, facteurs de transcription cellulaire. (qui envoient des messages erronés aux cellules rétiniennes) ou pour des éléments qui composent la structure des photorécepteurs. Les mutations génétiques héréditaires sont présentes dans les cellules dès la conception ; les anomalies courantes incluent celles des gènes RP1 (dans la rétinite pigmentaire-1, autosomique dominante) , RHO (RP4, autosomique dominante) et RDS (RP7, autosomique dominante) Les causes non héréditaires de rétinite pigmentaire sont rares, mais la possibilité de retrouver un cas isolé (mutation spontanée), dans lequel il n'y a pas d'antécédents familiaux de la maladie.