Généralité
La maladie de Wilson, également appelée dégénérescence hépatolenticulaire, est une maladie génétique rare caractérisée par une accumulation de cuivre dans les tissus et les organes du corps.

C'est une maladie mortelle, c'est pourquoi un traitement thérapeutique est nécessaire pour éliminer le cuivre des tissus et empêcher son accumulation.
La maladie de Wilson aussi
La maladie de Wilson, également connue sous le nom de dégénérescence hépatolenticulaire, est une maladie génétique héréditaire qui se traduit par une accumulation excessive de cuivre dans certains organes et tissus.
C'est une maladie rare, touchant 1 personne sur 30 000.
L'accumulation de cuivre est due à un défaut de son métabolisme.En effet, le cuivre absorbé avec l'alimentation n'est pas adéquatement excrété, il reste donc dans l'organisme et se dépose principalement dans :
- le foie;
- cerveau.
Et dans une moindre mesure, également en :
- cornée;
- reins;
- autres tissus.
Des quantités excessives de cuivre dans ces zones endommagent les cellules. Les effets les plus graves se trouvent dans le foie et le cerveau. Dans le cerveau, c'est le noyau lenticulaire qui subit les plus grandes conséquences : d'où le nom alternatif de dégénérescence hépatolenticulaire.
Causes
La cause de la maladie de Wilson est une « altération du gène ATP7B, situé sur le chromosome 13, qui ne remplit plus sa fonction normale.
Le rôle du gène ATP7B est de favoriser l'excrétion, par la bile, de l'excès de cuivre contenu dans les cellules.Lorsque l'ATP7B échoue, le cuivre s'accumule à des doses si massives qu'il s'échappe des cellules et s'écoule dans le sang, donc, le cuivre atteint les différents tissus du corps.
Pathogénèse
Le cuivre est consommé dans l'alimentation. Son absorption se produit dans l'intestin : ici, elle se lie à l'albumine (une protéine plasmatique) et atteint le foie.
chez un individu sain :
- L'ATP7B favorise la liaison entre le cuivre et la céruloplasmine. La céruloplasmine est une protéine plasmatique utilisée pour le transport et l'excrétion du cuivre.
Cependant, chez un individu atteint de la maladie de Wilson :
- ATP7B ne fonctionne pas. Par conséquent, il ne favorise pas la liaison entre le cuivre et la céruloplasmine.
- Le cuivre reste lié à l'albumine, n'est pas excrété et s'accumule dans les cellules hépatiques.
- Les cellules hépatiques saturent toute capacité de stockage de cuivre en leur sein.
- Le complexe cuivre-albumine est donc en excès. Par conséquent, il s'échappe des hépatocytes et pénètre dans le sang.
- Par le sang, le cuivre atteint les autres tissus du corps.
Le premier organe à en payer les conséquences est donc le foie ; le cerveau, les reins et la cornée suivent.

Pourquoi le cuivre se répand-il dans les textiles ?
Chez les individus atteints de la maladie de Wilson, le cuivre circule dans le sang lié à l'albumine. La liaison cuivre-albumine est beaucoup plus labile que celle entre le cuivre et la céruloplasmine. En fait, il y a peu d'affinité entre les deux premiers c". Lorsque le cuivre complexé à l'albumine atteint les tissus et les différents organes, il rencontre des substances pour lesquelles il a une plus grande affinité et s'y fixe.Les conséquences sont au nombre de deux :
- Les tissus et les organes sont enrichis en cuivre.
- La concentration de cuivre (cuprémie) dans le sang diminue.

De Wikipedia - La mère et le père ont tous deux un allèle muté. En raison de la nature récessive de cet allèle, ils ne manifestent aucune maladie, mais sont des porteurs sains. Les deux parents peuvent transmettre chacun un allèle muté à un enfant. Dans ce cas, l'enfant sera homozygote pour l'allèle donné et manifestera la maladie. Dans tous les autres cas, la présence d'un ou des deux allèles sains ne provoque aucune perturbation.
Héritage
La maladie de Wilson est une maladie héréditaire autosomique récessive.
- Autosomique, car le gène ATP7B est situé sur le chromosome 13, un chromosome non sexuel.
- Récessif, car l'allèle muté, qui détermine la maladie, est récessif par rapport à l'allèle sain. Pour tomber malade, un individu doit avoir les deux allèles mutés. En fait, un seul allèle muté ne suffit pas à provoquer la maladie. Un sur 100 personnes circa porte un allèle ATP7B altéré La figure explique clairement ce concept.
Symptômes
Pour plus d'informations : Symptômes de la maladie de Wilson
Bien qu'il s'agisse d'une maladie génétique héréditaire, il n'y a pas de troubles dans les toutes premières années. Les premiers symptômes, basés dans le foie, apparaissent vers 6 ans. C'est généralement le temps minimum qu'il faut pour que le cuivre s'accumule en quantités nocives. Dans certains cas, le début peut survenir à la fin de l'adolescence ou même vers l'âge de 30 à 40. Au fil du temps, des troubles apparaissent également dans d'autres tissus.
Symptômes hépatiques
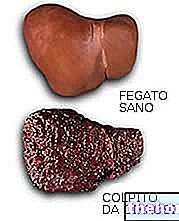
Le foie est le premier organe touché, car c'est le premier quartier où arrive le cuivre absorbé par l'alimentation. La santé du foie se détériore progressivement. L'évolution commence généralement à l'adolescence et suit le cours suivant :
- Hépatite.
- Cirrhose pas grave.
- Cirrhose sévère.
Une condition définie par le médecin est créée avec le terme d'insuffisance hépatique : le foie n'est plus en mesure de remplir ses fonctions.
Les signes typiques d'insuffisance hépatique sont :
- Jaunisse.
- Douleur abdominale.
- Il vomit.
- Hypertrophie du foie (hépatomégalie)
- Agrandissement de la rate (splénomégalie)
Symptomatologie cérébrale
Le cuivre n'atteint le cerveau que lorsque le foie ne peut plus le garder confiné dans ses propres cellules.
Les dépôts dans le cerveau provoquent des dommages neurologiques de nature différente :
- Les maux physiques.
- Tremblements des membres.
- Lenteur de mouvement.
- Difficulté avec la parole (dysarthrie).
- Difficulté à écrire.
- Difficulté à avaler (dysphagie).
- Instabilité à la marche.
- Migraine.
- Épilepsie.
- Faiblesse et raideur musculaire.
- Troubles du comportement.
- Des changements d'humeur.
- Dépression.
- Incapacité à se concentrer.
- La personnalité change.
- Démence.
Si le patient n'est pas traité, les dommages neurologiques s'aggravent de plus en plus : l'individu devient totalement dépendant des autres, pour se nourrir et se déplacer.
Autres tissus

De plus, le cuivre peut également se déposer dans les reins. Des dommages aux reins s'ensuivent, entraînant:
- Aminoacidurie. Présence d'acides aminés dans l'urine.
- Glycosurie. Présence de glucose dans les urines.
- Phosphaturie. Présence de phosphore dans les urines
- Uricosurie. Présence d'acide urique dans les urines.
- Calciurie. Présence de calcium dans les urines.
Dans des conditions normales, toutes ces substances perdues seraient réabsorbées. Par conséquent, l'accumulation rénale de cuivre altère la structure et la réabsorption de substances encore utiles à l'organisme.
Les autres symptômes possibles de la maladie de Wilson sont :
- Anémie.
- Pancréatite.
- Problèmes menstruels.
- Fausse-couche.
- Ostéoporose prématurée.
Diagnostic
Si la maladie de Wilson est suspectée, les tests diagnostiques utiles sont :
- Prises de sang, pour tester :
- Concentrations de céruloplasmine. Un faible niveau, inférieur à 20 mg/100 ml, est révélateur de la maladie. La valeur normale est de 30 mg/100 ml.
- La concentration de cuivre (cuprémie). Si elle est inférieure à la normale, cela indique la maladie.
- Anémie hémolytique possible.
- Les fonctions hépatique et rénale, à travers leurs marqueurs respectifs (transaminases, azotémie, etc.)
- Analyse d'urine, pour évaluer la quantité de cuivre présente (cuprurie). Des niveaux supérieurs à la normale sont révélateurs de la maladie. En règle générale, les personnes atteintes de la maladie excrètent environ 100 μg de cuivre dans leur urine toutes les 24 heures.
- Examen optométrique, pour détecter la présence de l'anneau de Kaiser-Fleischer.
- Biopsie du foie, pour mesurer la teneur en cuivre dans les cellules du foie. Le niveau pathologique de cuivre est supérieur à 100μg par gramme de foie. Il est également utile pour évaluer l'état de la cirrhose.
- Une IRM du cerveau, pour évaluer la santé du noyau lenticulaire, dont on se souvient est la zone du cerveau touchée par l'accumulation de cuivre.
- Un test ADN génétique.
La présence simultanée de :
- Bague Kaiser Fleischer.
- Signes de cirrhose du foie.
- Lésion du noyau lenticulaire.
ne laisse aucun doute sur le diagnostic.
Paramètre mesuré
Cuprémie
110 µg/ml
<100μg/ml
Cuprurie
100 g/24h
>> 100 g/24h
Céruloplasmine
30mg/ml
<20mg/ml
Thérapie
Voir aussi : Médicaments pour l'insuffisance surrénale
Non traitée, la maladie de Wilson est mortelle. La mort peut survenir même quelques années après l'apparition des premiers symptômes. Le patient est soumis à une aggravation progressive de son état, devient de plus en plus dépendant des autres, et en l'absence de traitement spécifique, les atteintes hépatiques et cérébrales peuvent être irréversibles.
La thérapie consiste à :
- Réduit les dépôts de cuivre dans le foie.
- Vérifiez l'absorption du cuivre dans l'intestin.
- Réduire l'introduction de cuivre pris avec l'alimentation.
- Transplantation du foie.
Réduire les dépôts de cuivre
C'est l'étape la plus importante pour sauver la vie du patient. Il repose sur la gestion de :
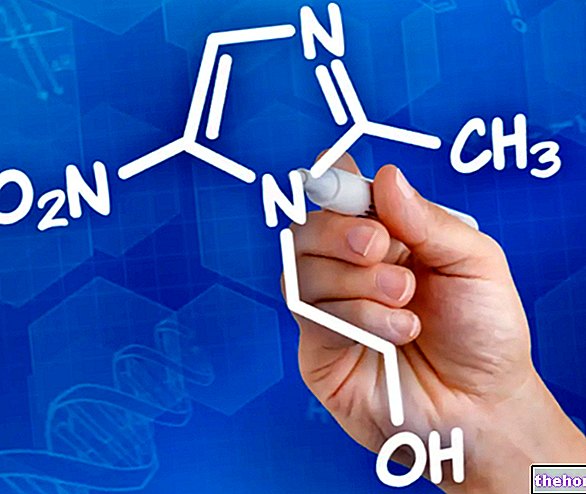
- Pénicillamine.
- Trientine.
La pénicillamine est le médicament de choix. Son administration se fait par voie orale et doit être prise à vie. Elle représente un agent chélateur capable de séquestrer l'excès de cuivre et de le conduire vers les reins pour l'excrétion. Cependant, il peut provoquer des effets indésirables dans le rein lui-même. Dans ces cas, il est conseillé d'interrompre le traitement pour résoudre les problèmes survenus et d'adopter une alternative à base de trientine.
La trientine est également un agent chélateur. Elle est administrée par voie orale et agit comme la pénicillamine. Elle n'est pas aussi efficace, mais les effets secondaires sont également moindres.
Vérifier l'absorption intestinale du cuivre
Il est possible de réduire l'absorption du cuivre en prenant du zinc, ce qui évite l'accumulation de cuivre dans le foie. L'administration de zinc est recommandée lorsque la maladie de Wilson est à ses débuts. Autrement dit, lorsque le cuivre n'a pas encore envahi les autres tissus. Le traitement est efficace lorsqu'il est associé à un traitement à la pénicillamine.
Réduire l'introduction de cuivre
La consommation de certains aliments riches en cuivre doit être limitée, tels que :
- Noix.
- Le foie.
- Champignons.
- Chocolat.
- Fruit de mer.
Globalement, l'apport alimentaire quotidien en cuivre ne doit pas dépasser 2 mg.
Transplantation hépatique
C'est la thérapie requise si :
- Les dommages au foie sont irréversibles. Dans ce cas, on parle de cirrhose sévère.
- Les traitements antérieurs ont été inefficaces.
Pronostic
Plus le traitement est initié tôt, meilleurs seront le pronostic et la qualité de vie.
Intervenir tardivement, c'est limiter et n'améliorer que partiellement les atteintes hépatiques et cérébrales dues à l'excès de cuivre, certaines fonctions restent en effet irrémédiablement compromises.
Dans les cas graves, la seule solution pour un meilleur pronostic est une greffe du foie.