Qu'est-ce que la phénylcétonurie
Là phénylcétonurie (P.K.U.) il s'agit d'une maladie métabolique héréditaire autosomique récessive qui affecte 1 individu sur 10 000 et semble survenir davantage chez les homozygotes que chez les hétérozygotes.
Appartenant au groupe des hyperphénylalaninémies, la phénylcétonurie compromet significativement le métabolisme de la phénylalanine et en particulier son conversion en tyrosine; la phénylcétonurie est reconnue par les taux urinaires élevés de phénylalanine et de certains dérivés (phénylpyruvate, phénylacétate, phénylactate et phénylacétylglutamine).
La complication la plus grave de la phénylcétonurie est la retard mental.
Phénylalanine, tyrosine et dérivés
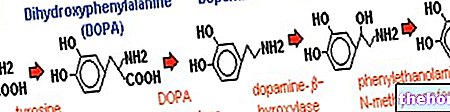
La phénylalanine est un acide aminé essentiel et constitue la majorité des protéines alimentaires ; il peut être converti par l'enzyme phénylalanine hydroxylase en tyrosine (en ajoutant un groupe hydroxyle -OH). À son tour, la tyrosine est un acide aminé précurseur pour la synthèse de :
- L-DOPA (intermédiaire de synthèse de la dopamine)
- Épinéphrine
- Norépinéphrine (tous les neurotransmetteurs).

Mécanisme de la phénylcétonurie (P.K.U.)
Comme anticipé, dans la phénylcétonurie, due à une ou plusieurs (6 au total) mutations chromosomiques, l'expression (donc l'activité métabolique) de la phénylalanine hydroxylase est pratiquement nulle. Ces altérations peuvent être de diverses natures (des changements "faux-sens" aux défauts "d'épissage" ou même aux "délétions partielles") mais ce qui compte c'est qu'en raison de cette inefficacité enzymatique les taux sanguins de phénylalanine (qui sont normalement de 1mg/100ml) dans la phénylcétonurie DOMINANTE ils atteignent facilement des quantités même 50 fois supérieures.
Fonctionnement de l'enzyme phénylalanine hydroxylase: Pour produire de la tyrosine (+ dihydrobioptérine), la phénylalanine hydroxylase nécessite : de la phénylalanine, de l'oxygène et de la tétrahydrobioptérine (une ptéridine réduite qui agit comme cofacteur) ; la réaction est également réversible et la dihydrobioptérine peut être reconvertie (grâce à l'enzyme dihydroptérine réductase) dans la tétrahydrobioptérine.
Complications
La phénylcétonurie peut donner lieu à des complications plus ou moins sévères selon la gravité de la manifestation pathologique et l'opportunité du diagnostic ; étant une pathologie héréditaire, la phénylcétonurie se distingue par :
- Dominante, donc caractérisée par une inactivité COMPLÈTE de l'enzyme phénylalanine hydroxylase
- Récessif, dans lequel seulement 30% du patrimoine enzymatique total est actif.
Les complications de la phénylcétonurie sont attribuables, et directement proportionnelles, à l'accumulation métabolique de la phénylalanine, de ses dérivés et à la synthèse réduite de la tyrosine.En pathologie, l'excès de phénylalanine est filtré relativement efficacement par le rein qui ne la réabsorbe que partiellement, l'éliminant avec les urines. ; cependant, la persistance des niveaux d'hyper-phénylalaninémie détermine une réaction métabolique de CONVERSION moléculaire chez acide phénylpyruvique et/ou d'autres dérivés plus faciles à drainer (phénylpyruvate, phénylacétate, phénylactate).
Ce qui complique la phénylcétonurie est la toxicité de la phénylalanine, de l'acide phénylpyruvique et de ses dérivés vis-à-vis du système nerveux central (SNC), leur présence excessive dans le développement cérébral détermine inexorablement une forme de retard mental.
NB. Les concentrations plasmatiques des autres acides aminés sont légèrement réduites, probablement en raison d'un retour sur l'absorption intestinale ou la réabsorption tubulaire rénale.
Les lésions cérébrales, en tant que complication grave de la phénylcétonurie, sont causées par la soustraction d'autres acides aminés essentiels dans la protéosynthèse, en particulier dans la formation de polyribosomes, de myéline, de noradrénaline et de sérotonine. La phénylcétonurie - non visible immédiatement après la naissance mais après quelques années - si elle n'est pas traitée, nécessite l'hospitalisation de l'enfant et est totalement irréversible.
La phénylcétonurie avancée peut également être clairement visible à l'œil nu; les fortes concentrations de phénylalanine, inhibant l'enzyme tyrosinase, altèrent significativement la synthèse de mélanine en diminuant la pigmentation de la peau et des cheveux ; de plus, l'accumulation de phénylacétate dans les cheveux et la peau confère aux phénylcétonuriques une "odeur de souris" forte et désagréable.