Généralité
Le rétinoblastome (Rb) est une tumeur oculaire maligne qui se développe à partir des cellules de la rétine. Ce cancer peut survenir à tout âge, mais son apparition est plus fréquente pendant la petite enfance avant l'âge de cinq ans.

Le cancer de l'enfant est agressif : le rétinoblastome peut se propager aux ganglions lymphatiques, aux os ou à la moelle osseuse. Rarement, elle implique le système nerveux central (cerveau et moelle épinière).
Environ 90 % des enfants atteints de rétinoblastome ont un pronostic positif (probabilité de guérison), à condition que le diagnostic soit précoce et que le traitement soit commencé avant que le cancer ne se propage. Dans la mesure du possible, le but de l'intervention médicale est de préserver la vision du patient.
Causes
La série d'événements conduisant à l'apparition d'une tumeur est complexe et commence lorsque les cellules de la rétine développent une mutation (ou délétion), impliquant le gène suppresseur de tumeur RB1, situé sur la bande q14 du chromosome 13 (13q14).
Chaque cellule a normalement deux gènes RB1:
- Si au moins une copie du gène fonctionne correctement, le rétinoblastome n'apparaît pas (mais le risque augmente) ;
- Lorsque les deux copies du gène sont mutées ou manquantes, une prolifération cellulaire incontrôlée se produit.
Dans de nombreux cas, on ne sait pas exactement ce qui induit des changements dans le gène RB1 (rétinoblastome sporadique) ; ceux-ci peuvent résulter d'erreurs génétiques aléatoires, qui se produisent, par exemple, lors de la reproduction et de la division cellulaire. Cependant, il est connu que les anomalies génétiques sous-jacentes au rétinoblastome peuvent également être transmises des parents aux enfants, avec un mode de transmission autosomique dominant. Cela signifie que si un parent est porteur d'un gène muté (dominant), chaque enfant aura 50 % de chance d'en hériter et 50 % de chance d'avoir une constitution génétique normale (gènes récessifs).
- Une cellule occasionnelle inactive sa seule copie normale du gène RB1 (une copie est déjà mutée) ;
- La perte des deux copies de RB1 entraîne une « prolifération excessive de la rétine.
- Une cellule occasionnelle inactive l'un de ses gènes RB1 normaux ;
- La deuxième copie du gène RB1 est inactivée ;
- La perte des deux copies de RB1 induit une prolifération cellulaire excessive qui conduit au rétinoblastome.
Caractéristiques génétiques et moléculaires
- Le rétinoblastome a été la première tumeur à être directement associée à une « anomalie génétique (délétion ou mutation de la bande q14 du chromosome 13).
- RB1 code pour la protéine pRb, qui joue un rôle clé dans le cycle cellulaire : elle permet la réplication de l'ADN et la progression du cycle cellulaire, car elle participe au contrôle de la transcription des gènes en phase S (G1 → † "S).
- En plus du rétinoblastome, le gène RB1 est inactivé dans les cancers de la vessie, du sein et du poumon.
Rétinoblastome héréditaire
Les enfants atteints de rétinoblastome héréditaire ont tendance à développer la maladie à un âge plus précoce que les cas sporadiques. De plus, ces enfants courent un risque accru d'autres cancers non oculaires, car l'anomalie du gène RB1 est congénitale (c'est-à-dire présente dès la naissance) et affecte toutes les cellules du corps (appelée mutation germinale), y compris celles des deux. rétines : Pour cette raison, les enfants atteints de la forme héréditaire ont souvent un rétinoblastome bilatéral plutôt qu'un seul œil.
Symptômes
Pour en savoir plus : Symptômes du rétinoblastome
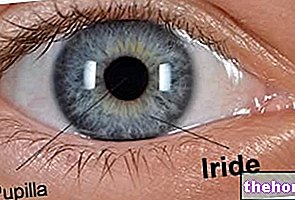
Le signe le plus courant et le plus évident du rétinoblastome est l'aspect anormal de la pupille, qui présente un reflet blanc grisâtre lorsqu'elle est touchée par un faisceau lumineux (leucocorie ou réflexe du chat amaurotique). Les autres signes et symptômes comprennent : une vision réduite, des douleurs et des rougeurs oculaires et un retard de développement. Certains enfants atteints de rétinoblastome peuvent développer un strabisme (yeux mal alignés); dans d'autres cas, il est possible de trouver un glaucome néovasculaire, qui, après un certain temps, peut provoquer une hypertrophie de l'œil (buftalmo).
Les cellules cancéreuses peuvent envahir davantage l'œil et d'autres structures :
- Rétinoblastome intraoculaire. Le rétinoblastome peut être défini comme intraoculaire lorsque la tumeur est entièrement située à l'intérieur de l'œil. Le néoplasme ne peut se trouver que dans la rétine ou affecter également d'autres parties, telles que la choroïde, le corps ciliaire et une partie du nerf optique. Par conséquent, le rétinoblastome intraoculaire ne se propage pas aux tissus autour de l'extérieur de l'œil.
- Rétinoblastome extraoculaire. La tumeur peut proliférer et affecter les tissus autour de l'œil (rétinoblastome orbitaire). Le cancer peut également se propager à d'autres régions du corps, telles que le cerveau, la colonne vertébrale, la moelle osseuse et les ganglions lymphatiques (rétinoblastome métastatique).
La présence d'une extension orbitaire, d'une atteinte de l'uvée et d'une invasion du nerf optique sont des facteurs de risque connus pour le développement d'un rétinoblastome métastatique.
Diagnostic
En cas d'antécédents familiaux positifs, le patient subit des examens oculaires réguliers pour le dépistage du cancer.Si le rétinoblastome congénital est bilatéral, il est généralement diagnostiqué dans la première année de vie, tandis que lorsqu'il n'affecte qu'un seul œil, la présence de la tumeur peut être confirmée vers 18-30 mois.
Le diagnostic clinique de rétinoblastome est établi par l'examen du fond d'œil.La tumeur, selon sa localisation, peut être visible lors d'un simple examen de l'œil, par ophtalmoscopie indirecte. Les techniques d'imagerie peuvent être utilisées pour confirmer le diagnostic, définir le stade de la tumeur (où elle se trouve, quelle est son étendue, si elle affecte les fonctions d'autres organes du corps, etc.) et déterminer si le traitement a été efficace . Les investigations peuvent inclure une échographie, une tomodensitométrie (TDM) et une imagerie par résonance magnétique (IRM).
Le diagnostic de génétique moléculaire est possible grâce à l'identification de la mutation du gène RB1 L'analyse cytogénétique (c'est-à-dire des chromosomes) des lymphocytes du sang périphérique permet de détecter des délétions ou des réarrangements impliquant le chromosome 13 (13q14.1-q14.2) .
Traitements
En cas de rétinoblastome, plusieurs options de traitement peuvent être utilisées.
Les objectifs du traitement sont :
- Éliminer la tumeur et sauver la vie du patient ;
- Sauvez l'œil si possible;
- Préserver la vision autant que possible ;
- Éviter le développement d'autres cancers, qui peuvent également être causés par un traitement, en particulier chez les enfants atteints de rétinoblastome héréditaire.
Le pronostic (probabilité de guérison) et les options de traitement dépendent des facteurs suivants :
- Stade de la tumeur ;
- L'âge et l'état de santé général du patient ;
- Emplacement, taille et nombre de foyers tumoraux ;
- Propagation du cancer à d'autres zones que le globe oculaire
- Quelle est la probabilité que la vision puisse être préservée dans un ou les deux yeux.
La plupart des cas de rétinoblastome sont diagnostiqués tôt et traités avec succès, avant que le cancer ne se métastase à l'extérieur du globe oculaire, ce qui entraîne un taux de guérison de plus de 90 %.