Généralité
L'ADN mitochondrial, ou ADNmt, est l'acide désoxyribonucléique qui réside à l'intérieur des mitochondries, c'est-à-dire les organites des cellules eucaryotes responsables du très important processus cellulaire de phosphorylation oxydative.

Cependant, il présente également certaines particularités, à la fois structurelles et fonctionnelles, qui le rendent unique en son genre. Ces particularités incluent : la circularité du double brin de nucléotides, le contenu des gènes (qui n'est que de 37 éléments) et l'absence quasi totale de séquences nucléotidiques non codantes.
L'ADN mitochondrial remplit une fonction fondamentale pour la survie des cellules : il produit les enzymes nécessaires à la réalisation de la phosphorylation oxydative.
Qu'est-ce que l'ADN mitochondrial ?
L'ADN mitochondrial, ou ADNmt, est l'ADN situé dans les mitochondries.
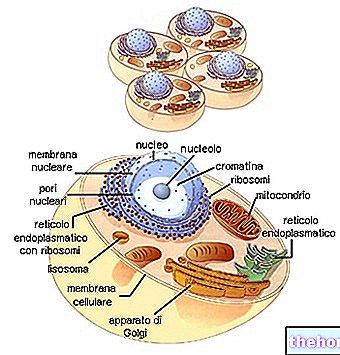
Les mitochondries sont ces grands organites cellulaires, typiques des organismes eucaryotes, qui convertissent l'énergie chimique contenue dans les aliments en ATP, qui est une forme d'énergie qui peut être exploitée par les cellules.
CONTEXTE SUR LA STRUCTURE ET LE FONCTIONNEMENT DES MITOCHONDRONES
De forme tubulaire, filamenteuse ou granuleuse, les mitochondries résident dans le cytoplasme, occupant près de 25 % du volume de ce dernier.
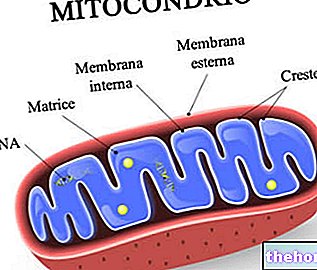
Ils ont deux membranes bicouches phospholipidiques, une plus externe et une plus interne.
La membrane la plus externe, connue sous le nom de membrane mitochondriale externe, représente le périmètre de chaque mitochondrie et possède des protéines de transport (porines et plus), qui la rendent perméable aux molécules d'une taille égale ou inférieure à 5 000 daltons.
La membrane la plus interne, connue sous le nom de membrane mitochondriale interne, contient tous les composants enzymatiques (ou enzymatiques) et coenzymes, nécessaires à la synthèse de l'ATP, et définit un espace central, appelé la matrice.
Contrairement à la membrane la plus externe, la membrane mitochondriale interne présente de nombreuses invaginations - appelées crêtes - qui augmentent sa surface totale.
Entre les deux membranes mitochondriales, il y a un espace de près de 60-80 Angströms (A).Cet espace est appelé l'espace intermembranaire.L'espace intermembranaire a une composition très similaire à celle du cytoplasme.
La synthèse d'ATP, opérée par les mitochondries, est un processus très complexe, que les biologistes identifient au terme de phosphorylation oxydative.
EMPLACEMENT PRÉCIS DE L'ADN MITOCHONDRAL ET QUANTITÉ

Figure : une mitochondrie humaine.
L'ADN mitochondrial réside dans la matrice mitochondriale, c'est-à-dire dans l'espace délimité par la membrane mitochondriale interne.
Selon des études scientifiques fiables, chaque mitochondrie peut contenir de 2 à 12 copies d'ADN mitochondrial.
Étant donné que, dans le corps humain, certaines cellules peuvent contenir plusieurs milliers de mitochondries, le nombre total de copies d'ADN mitochondrial dans une seule cellule humaine peut atteindre 20 000 unités.
Veuillez noter: le nombre de mitochondries dans les cellules humaines varie selon le type cellulaire. Par exemple, les hépatocytes (c'est-à-dire les cellules du foie) peuvent contenir entre 1 000 et 2 000 mitochondries chacun, tandis que les érythrocytes (c'est-à-dire les globules rouges) en sont totalement dépourvus.
Structure
La structure générale d'une molécule d'ADN mitochondrial ressemble à la structure générale de l'ADN nucléaire, c'est-à-dire au patrimoine génétique présent dans le noyau des cellules eucaryotes.
En fait, de manière analogue à l'ADN nucléaire:
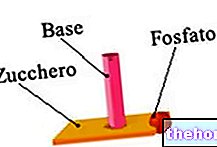
- L'ADN mitochondrial est un biopolymère, composé de deux longs brins de nucléotides. Les nucléotides sont des molécules organiques, résultant de l'union de trois éléments : un sucre à 5 atomes de carbone (dans le cas de l'ADN, le désoxyribose), une base azotée et un groupement phosphate.
- Chaque nucléotide de l'ADN mitochondrial se lie au nucléotide suivant du même brin au moyen d'une liaison phosphodiester entre le carbone 3 de son désoxyribose et le groupe phosphate du nucléotide suivant immédiatement.
- Les deux brins d'ADN mitochondrial ont des orientations opposées, l'extrémité de l'un interagissant avec l'extrémité de l'autre et vice versa.Cet arrangement particulier est connu sous le nom d'arrangement antiparallèle (ou orientation antiparallèle).
- Les deux brins d'ADN mitochondrial interagissent entre eux par l'intermédiaire des bases azotées.
En effet, chaque base azotée de chaque filament établit des liaisons hydrogène avec une et une seule base azotée, présente sur l'autre filament.
Ce type d'interaction est appelé « appariement entre bases azotées » ou « couple de bases azotées ». - Les bases azotées de l'ADN mitochondrial sont l'adénine, la thymine, la cytosine et la guanine.
Les appariements auxquels donnent lieu ces bases azotées ne sont pas aléatoires, mais très spécifiques : l'adénine n'interagit qu'avec la thymine, tandis que la cytosine n'interagit qu'avec la guanine. - L'ADN mitochondrial abrite des gènes (ou des séquences de gènes). Les gènes sont des séquences de nucléotides plus ou moins longs, avec une signification biologique bien définie. Dans la plupart des cas, ils donnent naissance à des protéines.

PARTICULARITÉS STRUCTURELLES DE L'ADN MITOCHONDRAL
Au-delà des analogies susmentionnées, l'ADN mitochondrial humain présente certaines particularités structurelles, qui le distinguent considérablement de l'ADN nucléaire humain.
Premièrement, c'est une molécule circulaire, tandis que l'ADN nucléaire est une molécule linéaire.
Ainsi, il possède 16 569 paires de bases azotées, tandis que l'ADN nucléaire en compte 3,3 milliards.
Il contient 37 gènes, tandis que l'ADN nucléaire semble en contenir entre 20 000 et 25 000.
Il n'est pas organisé en chromosomes, alors que l'ADN nucléaire est divisé en 23 chromosomes et forme, avec certaines protéines spécifiques, une substance appelée chromatine.
Enfin, il comprend une série de nucléotides qui participent à deux gènes à la fois, alors que l'ADN nucléaire possède des gènes dont les séquences nucléotidiques sont bien définies et distinctes les unes des autres.
Source
L'ADN mitochondrial a très probablement une origine "bactérienne".
En effet, sur la base de nombreuses études indépendantes, les biologistes moléculaires pensent que la présence cellulaire d'ADN mitochondrial est le résultat de l'incorporation, par des cellules eucaryotes ancestrales, d'organismes bactériens indépendants, très proches des mitochondries.
Cette curieuse découverte n'a que partiellement étonné la communauté scientifique, car l'ADN présent dans les bactéries est généralement un brin nucléotidique circulaire, comme l'ADN mitochondrial.
La théorie selon laquelle les mitochondries et l'ADN mitochondrial ont une « origine bactérienne prend le nom de « théorie endosymbiotique », du mot « endosymbiose ». Brièvement, en biologie, le terme « endosymbiose » désigne une collaboration entre deux organismes, qui implique la « incorporation de l'un dans l'autre, afin d'obtenir un certain avantage.
Curiosité
Selon des études scientifiques fiables, au cours de l'évolution de nombreux gènes bactériens, présents sur le futur ADN mitochondrial, auraient changé de localisation, se déplaçant dans l'ADN nucléaire.
En d'autres termes, au début de l'endosymbiose, certains gènes maintenant présents sur l'ADN nucléaire résidaient dans l'ADN de ces organismes bactériens, qui deviendraient plus tard des mitochondries.
Pour étayer la théorie relative à un déplacement des gènes entre l'ADN mitochondrial et l'ADN nucléaire, il y a l'observation que certains gènes dérivent de l'ADN mitochondrial, dans certaines espèces, et de l'ADN nucléaire, dans d'autres.
Fonction
L'ADN mitochondrial produit des enzymes (c'est-à-dire des protéines), nécessaires à la bonne mise en œuvre du délicat processus de phosphorylation oxydative.
Les instructions pour synthétiser ces enzymes résident dans les 37 gènes qui composent le génome de l'ADN mitochondrial.
QUEL CODE DES GÈNES DE L'ADN MITOCHONDRAL : LES DÉTAILS
Les 37 gènes de l'ADN mitochondrial codent pour : les protéines, l'ARNt et l'ARNr.
En particulier:
- 13 codent pour 13 protéines responsables de la phosphorylation oxydative
- 22 code pour 22 molécules d'ARNt
- 2 encodent 2 molécules d'ARNr
Les molécules d'ARNt et d'ARNr sont fondamentales pour la synthèse des 13 protéines susmentionnées, car elles constituent la machinerie qui régule leur production.
Donc, en d'autres termes, l'ADN mitochondrial possède l'information pour produire un certain ensemble de protéines et les outils nécessaires à leur synthèse.
Que sont l'ARN, l'ARNt et l'ARNr?
L'ARN, ou acide ribonucléique, est l'acide nucléique qui joue un rôle fondamental dans la génération des protéines, à partir de l'ADN.
Généralement monocaténaire, l'ANN peut exister sous diverses formes (ou types), selon la fonction spécifique à laquelle il est délégué.
L'ARNt et l'ARNr sont deux de ces formes possibles.
L'ARNt est utilisé pour ajouter des acides aminés au cours du processus de fabrication des protéines.Les acides aminés sont les unités moléculaires qui composent les protéines.
L'ARNr forme les ribosomes, c'est-à-dire les structures cellulaires dans lesquelles s'effectue la synthèse des protéines.
Pour connaître en détail l'ANN et ses fonctions, les lecteurs peuvent cliquer ici.
DÉTAILS FONCTIONNELS DE L'ADN MITOCHONDRAL
D'un point de vue fonctionnel, l'ADN mitochondrial présente des caractéristiques particulières qui le distinguent clairement de l'ADN nucléaire.
Voici en quoi consistent ces caractéristiques particulières :
- L'ADN mitochondrial est semi-indépendant, en ce sens qu'il nécessite l'intervention de certaines protéines synthétisées à partir de l'ADN nucléaire.
D'autre part, l'ADN nucléaire est complètement autonome et produit par lui-même tout ce dont il a besoin pour accomplir correctement ses tâches. - L'ADN mitochondrial a un code génétique légèrement différent de celui de l'ADN nucléaire. Cela conduit à un certain nombre de différences dans la fabrication des protéines : si une certaine séquence de nucléotides dans l'ADN nucléaire conduit à la création d'une certaine protéine, la même séquence dans l'ADN mitochondrial conduit à la formation d'une protéine légèrement différente.
- L'ADN mitochondrial a très peu de séquences nucléotidiques non codantes, c'est-à-dire qu'elles ne produisent aucune protéine, ARNt ou ARNr. En termes de pourcentage, seulement 3% de l'ADN mitochondrial est non codant.
D'un autre côté, l'ADN nucléaire n'est codant qu'à 7 %, il contient donc beaucoup de séquences nucléotidiques non codantes (jusqu'à 93 %).
Tableau : résumé des différences entre l'ADN mitochondrial humain et l'ADN nucléaire humain.
ADN mitochondrial
ADN nucléaire
- c'est circulaire
- c'est linéaire
- Il a 16 569 paires de bases azotées en tout
- Il a un total de 3,3 milliards de paires de bases azotées
- Il contient 37 gènes en tout
- Il contient entre 20 000 et 25 000 gènes
- Pour fonctionner correctement, il a besoin du support de certains produits géniques, dérivés de l'ADN nucléaire
- Il est autonome et produit par lui-même tout ce dont il a besoin pour bien remplir ses fonctions
- Il peut être présent en plusieurs exemplaires au sein de chaque mitochondrie individuelle
- Il est unique, c'est-à-dire qu'il est en un seul exemplaire et qu'il réside dans le noyau
- 97% de la séquence nucléotidique qui la compose est codante
- Seulement 7% de la séquence nucléotidique qui la compose est codante
- Il n'est pas organisé en chromosomes
- Il est divisé en 23 chromosomes
- Il utilise un code génétique légèrement différent de celui, pour ainsi dire, "traditionnel"
- Utiliser le code génétique "traditionnel"
- Son héritage est maternel
- Son héritage est moitié maternel et moitié paternel
- Certains de ses nucléotides participent à deux gènes en même temps
- Les séquences de nucléotides qui composent les gènes se distinguent bien les unes des autres
Héritage
L'hérédité de l'ADN mitochondrial est strictement maternelle.
Cela signifie que, dans un couple de parents, c'est la femme qui transmet l'ADN mitochondrial à la descendance (c'est-à-dire aux enfants).
À l'inverse de ce qui précède, l'hérédité de l'ADN nucléaire est à moitié maternelle et à moitié paternelle, c'est-à-dire que les deux parents contribuent à parts égales à la transmission de l'ADN nucléaire dans la progéniture.
Veuillez noter: l'hérédité maternelle de l'ADN mitochondrial implique également la structure mitochondriale. Par conséquent, les mitochondries présentes chez un individu sont maternelles.
Pathologies associées
Prémisse: Une mutation génétique est un changement permanent dans la séquence de nucléotides, qui constituent un gène d'ADN nucléaire ou mitochondrial.
Typiquement, la présence d'une mutation génétique entraîne une « altération ou une perte de la fonction normale du gène impliqué.
La présence de mutations dans les gènes de l'ADN mitochondrial peut entraîner un large éventail de maladies, notamment :
- Neuropathie optique héréditaire de Leber
- Syndrome de Kearns-Sayre
- syndrome de Leigh
- Le déficit en cytochrome C oxydase
- Ophtalmoplégie externe progressive
- Le syndrome de Pearson
- Encéphalomyopathie mitochondriale avec acidose lactique et épisodes de type accident vasculaire cérébral (syndrome MELAS)
- Diabète avec surdité transmise par la mère
- Épilepsie myoclonique avec fibres rouges irrégulières
Concernant les conditions pathologiques liées à une ou plusieurs mutations de l'ADN mitochondrial, deux aspects doivent être précisés.
Premièrement, la gravité de la maladie dépend de la relation quantitative entre les ADN mitochondriaux mutés et les ADN mitochondriaux sains et normaux. Si le nombre d'ADN mitochondriaux mutés est largement supérieur à celui d'ADN sains, l'affection qui en résulte sera plus grave.
Deuxièmement, les mutations de l'ADN mitochondrial n'affectent que certains tissus de l'organisme, en particulier ceux qui nécessitent de grandes quantités d'ATP résultant du processus de phosphorylation oxydative.C'est tout à fait compréhensible : subir plus d'un dysfonctionnement de l'ADN mitochondrial sont les cellules qui la fonction que remplit normalement l'ADN mitochondrial.
NEUROPATHIE OPTIQUE HÉRÉDITAIRE DE LEBER
La neuropathie optique héréditaire de Leber résulte de la mutation de jusqu'à quatre gènes de l'ADN mitochondrial. Ces gènes contiennent les informations qui conduisent à la synthèse du complexe I (ou NADH oxyde-réductase), l'une des différentes enzymes impliquées dans le processus de phosphorylation oxydative.
Les manifestations de la pathologie consistent en une dégénérescence progressive du nerf optique et une perte progressive de la vision.
SYNDROME DE KEARNS-SAYRE
Le syndrome de Kearns-Sayre apparaît en raison de l'absence d'une bonne quantité d'ADN mitochondrial (NB : l'absence d'une certaine séquence nucléotidique s'appelle une délétion).
Les personnes atteintes du syndrome de Kearns-Sayre développent une ophtalmoplégie (paralysie totale ou partielle des muscles oculomoteurs), une forme de rétinopathie et des anomalies du rythme cardiaque (bloc auriculo-ventriculaire).
LE SYNDROME DE LEIGH
Le syndrome de Leigh survient à la suite de mutations de l'ADN mitochondrial, qui peuvent affecter la protéine ATP-synthase (également appelée complexe V) et/ou certains ARNt.
Le syndrome de Leigh est une maladie neurologique évolutive, qui apparaît dans la petite enfance ou l'enfance et est responsable de : retard de développement, faiblesse musculaire, neuropathie périphérique, troubles moteurs, difficultés respiratoires et ophtalmoplégie.
DÉFICITÉ EN CYTOCHROME C OXIDASE
Le déficit en cytochrome C oxydase est dû à la mutation d'au moins 3 gènes de l'ADN mitochondrial. Ces gènes sont essentiels à la bonne synthèse de l'enzyme cytochrome C oxydase (ou complexe IV), impliquée dans le processus de phosphorylation oxydative.
Les manifestations typiques du déficit en cytochrome C oxydase consistent en : un dysfonctionnement des muscles squelettiques, un dysfonctionnement cardiaque, un dysfonctionnement rénal et un dysfonctionnement hépatique.
OPHTALMOPLEGIE EXTERNE PROGRESSIVE
L'ophtalmoplégie externe progressive résulte de l'absence d'un nombre important de nucléotides de l'ADN mitochondrial (délétion)
A caractère évolutif (comme le laisse deviner son nom), cette pathologie provoque une paralysie des muscles oculomoteurs, avec pour conséquence un ptosis et des troubles visuels considérables.
LE SYNDROME DE PEARSON
Le syndrome de Pearson apparaît suite à une délétion évidente de l'ADN mitochondrial, de manière similaire à l'ophtalmoplégie externe progressive et au syndrome de Kearns-Sayre.
Les manifestations typiques du syndrome de Pearson consistent en : anémie sidéroblastique, dysfonctionnement pancréatique (par exemple diabète insulino-dépendant), déficits neurologiques et troubles musculaires.
Le syndrome de Pearson provoque généralement la mort de la personne affectée à un jeune âge. En effet, les personnes touchées par cette pathologie atteignent rarement l'âge adulte.
SYNDROME DE MELAS
Le syndrome MELAS, également connu sous le nom d'encéphalomyopathie mitochondriale avec acidose lactique et épisodes de type accident vasculaire cérébral, résulte de la mutation d'au moins 5 gènes de l'ADN mitochondrial.
Ces gènes contribuent à la synthèse de la NADH oxyde-réductase, ou complexe I, et de certains ARNt.
Le syndrome MELAS implique la présence de troubles neurologiques, de troubles musculaires, d'une accumulation inhabituelle d'acide lactique dans les tissus (avec tous les symptômes qui l'accompagnent), de problèmes respiratoires, d'une perte de contrôle de la fonction intestinale, d'une fatigue récurrente, de problèmes rénaux, cardiaques, de diabète, d'épilepsie et manque de coordination.
AUTRES PATHOLOGIES
Selon diverses études scientifiques, des maladies telles que le syndrome des vomissements cycliques, la rétinite pigmentaire, l'ataxie, la maladie de Parkinson et la maladie d'Alzheimer, verraient également une implication de l'ADN mitochondrial et de certaines de ses mutations.