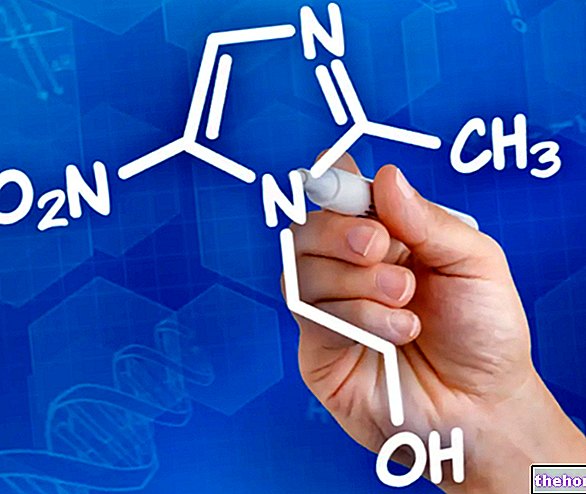
Ingrédients actifs : Adalimumab
Humira 40 mg solution injectable en stylo prérempli
Les notices d'emballage d'Humira sont disponibles pour les tailles de conditionnement :- Humira 40 mg/0,8 ml solution injectable à usage pédiatrique
- Humira 40 mg solution injectable en seringue préremplie
- Humira 40 mg solution injectable en seringue préremplie avec dispositif de sécurité pour aiguille
- Humira 40 mg solution injectable en stylo prérempli
Pourquoi Humira est-il utilisé ? Pourquoi est-ce?
Humira contient le principe actif adalimumab, un agent immunosuppresseur sélectif. Humira est indiqué pour le traitement de la polyarthrite rhumatoïde, de l'arthrite juvénile idiopathique polyarticulaire, de l'arthrite associée à l'enthésite, de la spondylarthrite ankylosante, de la spondylarthrite axiale sans signe radiographique de spondylarthrite ankylosante, du rhumatisme psoriasique, du rhumatisme psoriasique suppuré, de la maladie de Crohn et de la rectocolite hémorragique. processus inflammatoire de ces maladies. Le principe actif, l'adalimumab, est un anticorps monoclonal humain produit par des cultures cellulaires. Les anticorps monoclonaux sont des protéines qui reconnaissent et se lient à d'autres protéines. L'adalimumab se lie à une protéine spécifique (facteur de nécrose tumorale ou TNFα) qui est présent à des niveaux élevés dans les maladies inflammatoires telles que la polyarthrite rhumatoïde, l'arthrite juvénile idiopathique polyarticulaire, l'arthrite associée à l'enthésite, la spondylarthrite ankylosante, la spondylarthrite axiale sans signe radiographique de spondylarthrite ankylosante, rhumatisme psoriasique, psoriasis, hidrosadénite suppurée, maladie de Crohn et rectocolite hémorragique.
La polyarthrite rhumatoïde
La polyarthrite rhumatoïde est une maladie inflammatoire des articulations.
Humira est utilisé pour traiter la polyarthrite rhumatoïde chez l'adulte. Si vous souffrez de polyarthrite rhumatoïde modérée à sévère, d'autres médicaments de fond tels que le méthotrexate peuvent être utilisés dans un premier temps. Si la réponse à ces médicaments n'est pas satisfaisante, vous recevrez Humira pour traiter la polyarthrite rhumatoïde.
Humira peut également être utilisé pour le traitement de la polyarthrite rhumatoïde sévère, active et évolutive sans traitement préalable par méthotrexate.
Il a été démontré que Humira ralentit la progression des dommages au cartilage et aux os dans les articulations causés par la maladie et améliore la fonction physique.
Humira est généralement utilisé avec le méthotrexate. Si votre médecin décide que le traitement par méthotrexate est inapproprié, Humira peut être administré seul.
Arthrite juvénile idiopathique polyarticulaire et arthrite associée à l'enthésite
L'arthrite juvénile idiopathique polyarticulaire et l'arthrite associée à l'enthésite sont des maladies inflammatoires.
Humira est utilisé pour traiter l'arthrite juvénile idiopathique polyarticulaire chez les enfants et les adolescents âgés de 2 à 17 ans et l'arthrite associée à l'enthésite chez les enfants et les adolescents âgés de 6 à 17 ans. D'autres médicaments modificateurs de la maladie, tels que le méthotrexate, peuvent être administrés au moment du diagnostic. Si la réponse à ces médicaments n'est pas adéquate, Humira vous sera administré pour le traitement de l'arthrite juvénile idiopathique polyarticulaire ou de l'arthrite associée à l'enthésite.
Spondylarthrite ankylosante et spondylarthrite axiale sans signe radiographique de spondylarthrite ankylosante
La spondylarthrite ankylosante et la spondylarthrite axiale sans signe radiographique de spondylarthrite ankylosante sont des inflammations de la colonne vertébrale.
Humira est utilisé pour traiter la spondylarthrite ankylosante et la spondylarthrite axiale sans signe radiographique de spondylarthrite ankylosante chez l'adulte. Si vous souffrez de spondylarthrite ankylosante ou de spondylarthrite axiale sans signe radiographique de spondylarthrite ankylosante, vous prendrez d'abord d'autres médicaments. Si vous ne parvenez pas à obtenir une réponse adéquate avec ces médicaments, vous prendrez Humira pour réduire les signes et symptômes de la maladie.
Arthrite psoriasique
Le rhumatisme psoriasique est une inflammation des articulations associée au psoriasis. Humira est utilisé pour traiter le rhumatisme psoriasique chez l'adulte.
Il a été démontré que Humira ralentit les dommages au cartilage et aux os dans les articulations causés par la maladie et améliore la fonction physique.
Psoriasis en plaques chez l'adulte et l'enfant
Le psoriasis en plaques est une affection cutanée qui provoque des plaques de peau rougeâtres, squameuses et durcies recouvertes d'écailles argentées. On pense que le psoriasis est causé par un problème avec le système immunitaire du corps qui entraîne une augmentation de la production de cellules de la peau.
Humira est utilisé pour traiter le psoriasis en plaques modéré à sévère chez l'adulte. Si vous êtes un adulte atteint de psoriasis en plaques modéré à sévère, vous prendrez d'abord d'autres médicaments ou subirez une photothérapie. Si vous ne développez pas une réponse satisfaisante à ces traitements, vous recevrez Humira pour réduire les signes et symptômes du psoriasis.
Humira est également utilisé pour traiter le psoriasis en plaques sévère chez les enfants et les adolescents âgés de 4 à 17 ans chez qui le traitement topique et la photothérapie n'ont pas fonctionné de manière optimale ou ne sont pas indiqués.
Hydradénite suppurée
L'hidrosadénite suppurée (parfois appelée acné inversa) est une maladie inflammatoire chronique de la peau et est souvent douloureuse. Les symptômes peuvent inclure des bosses douloureuses et des abcès (kystes) qui peuvent drainer le pus. Le plus souvent, elle affecte des zones spécifiques de la peau, comme la région des aisselles. , les aisselles, l'intérieur des cuisses, l'aine et les fesses. Des cicatrices peuvent également se former dans les zones touchées.
Humira est utilisé pour traiter l'hidrosadénite suppurée chez l'adulte. Humira peut réduire le nombre de grumeaux et d'abcès que vous avez, ainsi que la douleur qui est souvent associée à cette maladie.
La maladie de Crohn chez l'adulte et l'enfant
La maladie de Crohn est une « inflammation du tube digestif.
Humira est utilisé pour traiter la maladie de Crohn chez les adultes et les enfants âgés de 6 à 17 ans. Si vous souffrez de la maladie de Crohn, vous recevrez d'abord d'autres médicaments. Si vous ne répondez pas suffisamment à ces médicaments, vous recevrez Humira pour réduire les symptômes typiques de la maladie de Crohn.
Rectocolite hémorragique
La rectocolite hémorragique est une « inflammation de l'intestin ».
Humira est utilisé pour traiter la rectocolite hémorragique chez l'adulte. Si vous souffrez de colite ulcéreuse, vous prendrez d'abord d'autres médicaments. Si vous ne parvenez pas à obtenir une réponse adéquate avec ces médicaments, vous prendrez Humira pour réduire les signes et symptômes de la maladie.
Contre-indications Quand Humira ne doit pas être utilisé
Ne pas utiliser Humira
- Si vous êtes allergique à l'adalimumab ou à l'un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
- Si vous avez une « infection grave, y compris une tuberculose active (voir « Avertissements et précautions »). Il est important d'informer votre médecin si vous présentez des signes ou des symptômes d'infection, tels que fièvre, plaies, sensation de fatigue, problèmes dentaires.
- En présence d'insuffisance cardiaque modérée ou sévère. Il est important d'informer votre médecin s'il y a eu ou s'il s'agit d'une maladie cardiaque grave (voir « Avertissements et précautions »).
Précautions d'emploi Quelles sont les informations à connaître avant de prendre Humira
Adressez-vous à votre médecin ou pharmacien avant d'utiliser Humira
- Si vous présentez des réactions allergiques accompagnées de symptômes tels qu'une oppression thoracique, une respiration sifflante, des étourdissements, un gonflement ou une éruption cutanée, arrêtez de prendre Humira et contactez immédiatement votre médecin.
- Si vous avez une infection, y compris des infections à long terme ou localisées (par exemple, des ulcères de jambe), consultez votre médecin avant de commencer le traitement par Humira. En cas de doute, contactez votre médecin.
- Vous pouvez contracter des infections plus facilement pendant votre traitement par Humira. Ce risque peut augmenter si votre fonction pulmonaire est compromise. Ces infections peuvent être graves et inclure la tuberculose, les infections causées par des virus, des champignons, des parasites ou des bactéries, ou d'autres infections opportunistes et la septicémie qui peuvent, dans de rares cas, mettre la vie en danger. Il est important d'informer votre médecin des symptômes tels que fièvre, plaies, fatigue ou problèmes dentaires. Votre médecin peut vous recommander d'arrêter temporairement Humira.
- Étant donné qu'il y a eu des cas de tuberculose chez des patients recevant Humira, votre médecin devra vérifier si vous présentez des signes ou des symptômes typiques de tuberculose avant de commencer le traitement par Humira. Cela impliquera la collecte d'une évaluation médicale détaillée qui comprend vos antécédents médicaux et les tests cliniques appropriés (par exemple, une radiographie pulmonaire et un test à la tuberculine). Les performances et les résultats de ces tests doivent être enregistrés dans l'alerte patient. Il est très important d'informer le médecin si vous avez déjà eu la tuberculose, ou si vous avez été en contact étroit avec des patients tuberculeux. La tuberculose peut survenir pendant le traitement malgré le fait que vous avez reçu un traitement préventif contre la tuberculose Contactez immédiatement votre médecin si des symptômes de tuberculose (toux persistante, perte de poids, apathie, fièvre modérée) ou d'autres infections apparaissent pendant ou après le traitement.
- Informez votre médecin si vous résidez ou voyagez dans des régions où les infections fongiques, telles que l'histoplasmose, la coccidioïdomycose ou la blastomycose, sont endémiques.
- Informez votre médecin si vous avez déjà eu des infections récurrentes ou si vous souffrez d'affections qui augmentent le risque d'infection.
- Informez votre médecin si vous êtes porteur du virus de l'hépatite B (VHB), si vous avez une infection active par le virus de l'hépatite B ou si vous pensez que vous pourriez être à risque de contracter le virus de l'hépatite B. vous devez subir un test de dépistage du virus de l'hépatite B. La prise d'Humira peut provoquer la réactivation du virus de l'hépatite B chez les personnes porteuses de ce virus. Dans certains cas rares, en particulier si le patient suit un traitement avec d'autres médicaments qui suppriment le système immunitaire, la réactivation du virus de l'hépatite B peut mettre la vie en danger.
- Si vous avez plus de 65 ans, vous pouvez être plus sensible aux infections pendant que vous prenez Humira. Vous et votre médecin devez porter une attention particulière aux signes d'infection pendant le traitement par Humira. Il est important d'informer votre médecin si des symptômes d'infections tels que fièvre, plaies , sensation de fatigue ou problèmes dentaires.
- Avant une intervention chirurgicale ou une intervention dentaire, informez votre médecin que vous prenez Humira. Votre médecin peut recommander une suspension temporaire.
- Si vous souffrez de maladies démyélinisantes telles que la sclérose en plaques, votre médecin décidera si vous devez commencer un traitement par Humira.
- Certains vaccins peuvent provoquer des infections et ne doivent pas être administrés pendant le traitement par Humira. Consultez votre médecin avant de vous faire vacciner. Chez les enfants, il est recommandé, si possible, de mettre en œuvre le calendrier vaccinal prévu, conformément aux directives de vaccination en vigueur, avant de commencer le traitement par Humira. Si vous avez pris Humira pendant votre grossesse, votre bébé peut avoir un risque accru de contracter cette infection jusqu'à environ 5 mois après la dernière dose que vous avez prise pendant la grossesse. Il est important que vous informiez votre pédiatre ou un autre professionnel de la santé. pendant la grossesse, afin qu'ils puissent décider quand votre bébé doit recevoir tout type de vaccination.
- En cas d'insuffisance cardiaque légère et de traitement concomitant par Humira, votre médecin devra évaluer et surveiller attentivement l'état de votre cœur. Il est important d'informer votre médecin de tout problème cardiaque, passé et présent. Si de nouveaux symptômes d'insuffisance cardiaque apparaissent ou si les symptômes existants s'aggravent (par exemple, essoufflement ou gonflement des pieds), contactez immédiatement votre médecin. Votre médecin décidera si vous pouvez prendre Humira.
- Chez certains patients, le corps peut ne pas être en mesure de produire suffisamment de cellules sanguines pour combattre l'infection ou arrêter les saignements. Si vous avez une fièvre persistante, des ecchymoses ou des saignements faciles ou une pâleur, contactez immédiatement votre médecin. Ce dernier peut décider d'arrêter le traitement.
- Certains types de cancers sont survenus très rarement chez des patients, enfants et adultes, recevant un traitement par Humira ou d'autres médicaments anti-TNF. Les patients atteints de polyarthrite rhumatoïde sévère à long terme peuvent avoir un risque plus élevé que la moyenne de développer un lymphome (un type de cancer qui affecte le système lymphatique) et une leucémie (un type de cancer qui affecte le sang et la moelle osseuse). Si vous prenez Humira, le risque de développer un lymphome, une leucémie ou d'autres cancers peut augmenter. Dans de rares circonstances, un type spécifique et sévère de lymphome a été observé chez les patients recevant Humira. Certains de ces patients étaient également sous traitement à l'azathioprine ou à la 6-mercaptopurine. Informez votre médecin si vous prenez de l'azathioprine ou de la 6-mercaptopurine avec Humira. De plus, des cas de cancer de la peau non mélanique ont été observés chez des patients prenant Humira. Si de nouvelles lésions cutanées apparaissent pendant ou après le traitement, ou si l'apparence des lésions existantes change, veuillez en informer votre médecin.
- Il y a eu des cas de tumeurs malignes, en plus du lymphome, chez des patients atteints d'un type spécifique de maladie pulmonaire appelée maladie pulmonaire obstructive chronique (MPOC) traités avec un autre anti-TNF. Si vous souffrez de MPOC ou si vous fumez beaucoup, vous devriez discuter avec votre médecin pour savoir si un traitement par un anti-TNF est approprié.
Enfants et adolescents
- Vaccinations : si possible, les enfants doivent avoir déjà été vaccinés avant d'utiliser Humira.
- Ne pas administrer Humira aux enfants de moins de 2 ans atteints d'arthrite juvénile idiopathique polyarticulaire.
Interactions Quels médicaments ou aliments peuvent modifier l'effet d'Humira
Autres médicaments et Humira
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Humira peut être pris avec du méthotrexate ou d'autres antirhumatismaux de fond (sulfasalazine, hydroxychloroquine, léflunomide et sels d'or parentéraux), des stéroïdes ou des analgésiques, y compris des anti-inflammatoires non stéroïdiens (AINS).
Humira ne doit pas être pris en même temps que des médicaments contenant de l'anakinra ou de l'abatacept comme principe actif. Si vous n'êtes pas sûr, demandez à votre médecin.
Humira avec de la nourriture et des boissons
Comme Humira est injecté sous la peau (par voie sous-cutanée), les aliments et les boissons n'interfèrent pas avec Humira.
Avertissements Il est important de savoir que :
La grossesse et l'allaitement
Les effets d'Humira chez la femme enceinte ne sont pas connus, c'est pourquoi l'utilisation d'Humira chez la femme enceinte n'est pas recommandée. Il est conseillé d'éviter une grossesse en utilisant une contraception adéquate pendant le traitement par Humira et pendant au moins 5 mois après le dernier traitement médicamenteux. Si vous tombez enceinte, vous devriez consulter votre médecin.
On ne sait pas si l'adalimumab passe dans le lait maternel.
Si vous allaitez, vous devez arrêter d'allaiter pendant le traitement par Humira et pendant au moins 5 mois après votre dernier traitement par Humira. Si vous avez pris Humira pendant la grossesse, votre bébé peut avoir un risque accru de contracter une infection.Il est important que vous informiez votre pédiatre ou un autre professionnel de la santé de votre utilisation d'Humira pendant la grossesse, avant que votre bébé ne reçoive tout type de vaccin. (pour plus d'informations, voir la section sur la vaccination).
Si vous suspectez ou envisagez de devenir enceinte, demandez conseil à votre médecin ou à votre pharmacien avant d'utiliser ce médicament.
Conduire et utiliser des machines
Humira peut affecter votre aptitude à conduire, à faire du vélo ou à utiliser des machines, même de manière modeste. Après avoir pris Humira, vous pouvez avoir des troubles visuels et avoir l'impression que votre environnement tourne.
Posologie et mode d'utilisation Comment utiliser Humira : Posologie
Utilisez toujours ce médicament en suivant exactement les indications de votre médecin ou pharmacien. En cas de doute, consultez votre médecin ou votre pharmacien
Adultes atteints de polyarthrite rhumatoïde, de rhumatisme psoriasique, de spondylarthrite ankylosante ou de spondylarthrite axiale sans signe radiographique de spondylarthrite ankylosante.
Humira est injecté sous la peau (voie sous-cutanée). La posologie habituelle chez les patients adultes atteints de polyarthrite rhumatoïde, de spondylarthrite ankylosante, de spondylarthrite axiale sans signe radiographique de spondylarthrite ankylosante et de rhumatisme psoriasique est de 40 mg d'adalimumab toutes les deux semaines, en dose unique.
Dans la polyarthrite rhumatoïde, le méthotrexate est poursuivi pendant le traitement par Humira.Si votre médecin décide que le méthotrexate est inapproprié, Humira peut être administré seul.
Si vous souffrez de polyarthrite rhumatoïde et que vous ne recevez pas de méthotrexate en association avec un traitement par Humira, votre médecin peut décider de vous prescrire 40 mg d'adalimumab par semaine.
Enfants atteints d'arthrite juvénile idiopathique polyarticulaire
La dose recommandée d'Humira pour les patients atteints d'arthrite juvénile idiopathique polyarticulaire âgés de 2 à 12 ans dépend de la taille et du poids de l'enfant.Le médecin de votre enfant vous conseillera sur la dose correcte à utiliser.
La dose recommandée d'Humira pour les patients atteints d'arthrite juvénile idiopathique polyarticulaire âgés de 13 à 17 ans est de 40 mg toutes les deux semaines.
Enfants atteints d'arthrite associée à une enthésite
La dose recommandée d'Humira pour les patients atteints d'arthrite associée à l'enthésite âgés de 6 à 17 ans dépend de la taille et du poids de l'enfant.
Adultes atteints de psoriasis
La dose habituelle d'Humira pour les adultes atteints de psoriasis est une dose initiale de 80 mg, suivie d'une dose de 40 mg, administrée toutes les deux semaines, en commençant la semaine suivant la dose initiale. Vous devez poursuivre le traitement par Humira aussi longtemps que votre le docteur vous dit.
Enfants ou adolescents atteints de psoriasis en plaques
La dose recommandée d'Humira pour les patients âgés de 4 à 17 ans atteints de psoriasis en plaques dépend du poids de l'enfant. Le médecin de votre enfant vous indiquera la dose correcte à utiliser. Les patients nécessitant une dose inférieure à 40 mg doivent utiliser Humira dans la présentation en flacon de 40 mg.
Adultes atteints d'hidradénite suppurée
La dose habituelle pour l'hidrosadénite suppurée est une dose initiale de 160 mg (4 injections en un jour ou 2 injections par jour pendant deux jours consécutifs), suivie d'une dose de 80 mg (2 injections le même jour) deux semaines plus tard. encore deux semaines, continuer avec une dose de 40 mg par semaine Il est recommandé d'utiliser quotidiennement une solution de lavage antiseptique sur les zones touchées.
Enfants ou adolescents atteints de la maladie de Crohn
Enfants ou adolescents pesant moins de 40 kg :
Le schéma posologique habituel est de 40 mg au début suivi de 20 mg deux semaines plus tard. Si une réponse plus rapide est requise, le médecin peut prescrire une dose initiale de 80 mg (en deux injections par jour) suivie de 40 mg deux semaines plus tard .
Par la suite, la dose habituelle est de 20 mg toutes les deux semaines. En fonction de la réponse de l'enfant, le médecin peut augmenter la fréquence de la dose à 20 mg chaque semaine.
Enfants ou adolescents pesant 40 kg ou plus :
Le schéma posologique habituel est de 80 mg au début suivi de 40 mg deux semaines plus tard. Si une réponse plus rapide est requise, le médecin peut prescrire une dose initiale de 160 mg (sous forme de 4 injections par jour ou de 2 injections par jour pendant 2 jours consécutifs) suivi de 80 mg deux semaines plus tard.
Par la suite, la dose habituelle est de 40 mg toutes les deux semaines. En fonction de la réponse de l'enfant, le médecin peut augmenter la fréquence des doses à 40 mg chaque semaine.
Les patients nécessitant une dose inférieure à 40 mg doivent utiliser Humira dans la présentation en flacon de 40 mg.
Adultes atteints de rectocolite hémorragique
La dose habituelle d'Humira pour les adultes atteints de rectocolite hémorragique est de 160 mg à la semaine 0 (la dose peut être administrée en quatre injections par jour ou en deux injections par jour pendant deux jours consécutifs) et égale à 80 mg par semaine.2, et puis à 40 mg toutes les deux semaines. En fonction de la réponse clinique, votre médecin peut augmenter la dose à 40 mg chaque semaine.
Mode et voie d'administration
Humira est administré par injection sous la peau (par injection sous-cutanée).
Instructions pour la préparation et l'injection d'Humira :
Les instructions ci-dessous vous montrent comment injecter Humira. Lisez attentivement les instructions et suivez-les étape par étape. Votre médecin ou son assistant vous renseignera sur la technique d'auto-administration.Ne vous injectez pas tant que vous n'êtes pas sûr de comprendre comment préparer et administrer l'administration. Après des instructions appropriées
Le contenu de la seringue ne doit pas être mélangé avec d'autres médicaments dans la même seringue ou flacon.
1) Préparation
- Lavez-vous soigneusement les mains.
- Placez les éléments suivants sur une surface propre : une seringue préremplie d'Humira pour injection et un tampon d'alcool.
- Vérifiez la date de péremption sur la seringue. Ne pas utiliser le produit au-delà du mois et de l'année indiqués.
2) Choix et préparation d'un site d'injection
- Choisissez un endroit sur la cuisse ou le ventre.
- Chaque nouvelle injection doit être effectuée à au moins 3 cm du site de la dernière injection. Ne pas injecter dans des zones où la peau est rouge, contusionnée ou dure. Cela peut indiquer une infection. Essuyez le site d'injection avec le tampon imbibé d'alcool en effectuant un mouvement de torsion. Ne touchez plus la zone avant l'injection.
3) Injection d'Humira
- NE PAS secouer la seringue.
- Retirez le capuchon de l'aiguille de la seringue en veillant à ne pas toucher l'aiguille ou à ce que l'aiguille ne touche aucune surface.
- Avec une main, prenez doucement la zone déjà frottée avec de l'alcool et maintenez-la immobile.
- Avec votre autre main, tenez la seringue (avec le côté nervuré vers le haut) à un angle de 45 ° par rapport au site d'injection.
- D'un mouvement ferme et rapide, enfoncez toute l'aiguille dans la peau.
- Laissez la peau avec la première main.
- Appuyez sur le piston pour injecter la solution - cela peut prendre 2 à 5 secondes pour vider la seringue.
- Lorsque la seringue est vide, retirez l'aiguille de votre peau en la gardant au même angle que lors de son introduction.
- Avec votre pouce ou un morceau de gaze, appuyez sur le site d'injection pendant 10 secondes. Un petit saignement peut survenir. Ne pas masser le site d'injection. Si vous le souhaitez, appliquez un patch.
Élimination des matériaux
- NE JAMAIS réutiliser la seringue Humira. Ne rebouchez JAMAIS l'aiguille.
- Après avoir injecté Humira, jetez immédiatement la seringue usagée dans un récipient spécial comme indiqué par votre médecin, infirmier/ère ou pharmacien.
- Gardez le récipient hors de la vue et de la portée des enfants.
Surdosage Que faire si vous avez pris trop d'Humira
Si vous avez utilisé plus d'Humira que vous n'auriez dû :
Si vous injectez accidentellement Humira plus fréquemment que ne vous l'a prescrit votre médecin ou votre pharmacien, contactez votre médecin ou votre pharmacien pour l'informer que vous avez pris plus de médicament. Conservez toujours la boîte à médicaments, même si elle est vide.
Si vous oubliez d'utiliser Humira :
Si vous oubliez de recevoir une injection, vous devez injecter votre prochaine dose d'Humira dès que vous vous en rendez compte, puis reprenez votre dose régulièrement selon votre horaire habituel.
Si vous arrêtez de prendre Humira
La décision d'arrêter l'utilisation d'Humira doit être discutée avec votre médecin.Les symptômes peuvent réapparaître après l'arrêt.
Si vous avez d'autres questions sur l'utilisation de ce médicament, demandez plus d'informations à votre médecin ou votre pharmacien.
Effets secondaires Quels sont les effets secondaires d'Humira
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, bien que tout le monde n'y soit pas sujet. La plupart des effets secondaires sont légers à modérés. Cependant, certains peuvent être graves et nécessiter un traitement. Des effets secondaires peuvent survenir jusqu'à 4 mois après la dernière injection d'Humira.
Informez immédiatement votre médecin si vous remarquez l'une des réactions suivantes :
- éruption cutanée sévère, urticaire ou autres signes d'une réaction allergique;
- gonflement du visage, des mains, des pieds;
- difficulté à respirer, difficulté à avaler;
- essoufflement à l'effort ou en position couchée ou pieds enflés.
Informez votre médecin dès que possible si vous remarquez l'une des réactions suivantes :
- signes d'infection tels que fièvre, malaise, plaies, problèmes dentaires, sensation de brûlure en urinant ;
- fatigue ou faiblesse;
- la toux;
- picotements;
- engourdissement;
- vision double;
- faiblesse des bras ou des jambes;
- gonflement ou plaie ouverte qui ne guérit pas
- signes et symptômes suggérant l'apparition de troubles affectant le système hématopoïétique, tels que la présence d'une fièvre persistante, des ecchymoses, des hémorragies, une pâleur.
Les symptômes décrits ci-dessus peuvent être des signes des effets indésirables suivants, qui ont été observés avec Humira :
Très fréquent (peut affecter plus de 1 personne sur 10) :
- réactions au site d'injection (y compris douleur, gonflement, rougeur ou démangeaison);
- infections des voies respiratoires (y compris rhumes, rhinorrhée, sinusite et pneumonie);
- mal de tête;
- douleur abdominale;
- nausées et vomissements;
- éruption;
- douleurs musculo-squelettiques.
Fréquent (peut affecter jusqu'à 1 personne sur 10) :
- infections graves (y compris septicémie et grippe);
- infections cutanées (y compris la cellulite et l'infection par le zona);
- infections de l'oreille;
- infections buccales (y compris les infections dentaires et l'herpès simplex);
- infections du système reproducteur;
- infections des voies urinaires;
- les infections fongiques;
- infections articulaires;
- tumeurs bénignes;
- cancer de la peau;
- réactions allergiques (y compris allergie saisonnière);
- déshydratation;
- changements d'humeur (y compris la dépression);
- anxiété;
- les troubles du sommeil;
- troubles de la sensibilité tels que picotements, contractions musculaires ou engourdissements ;
- migraine;
- compression des racines nerveuses (y compris les douleurs lombaires et les douleurs aux jambes);
- troubles visuels;
- inflammation des yeux;
- inflammation des paupières et gonflement des yeux;
- vertiges;
- sensation de rythme cardiaque rapide;
- hypertension;
- les bouffées de chaleur;
- hématome;
- la toux;
- asthme;
- essoufflement;
- saignement gastro-intestinal;
- dyspepsie (indigestion, ballonnements, brûlures d'estomac);
- reflux acide;
- syndrome sec (y compris sécheresse des yeux et de la bouche) ;
- démanger;
- démangeaisons;
- hématome;
- inflammation de la peau (telle que l'eczéma);
- rupture des ongles des doigts et des orteils;
- transpiration accrue;
- perte de cheveux;
- apparition ou aggravation du psoriasis;
- spasmes musculaires;
- sang dans les urines;
- problèmes rénaux;
- douleur thoracique;
- œdème;
- fièvre;
- réduction des plaquettes dans la sanggue qui augmente le risque de saignement ou d'ecchymose;
- difficulté à guérir.
Peu fréquent (peut affecter jusqu'à 1 personne sur 100) :
- les infections opportunistes (qui incluent la tuberculose et d'autres infections qui surviennent lorsque les défenses immunitaires sont réduites) ;
- infections neurologiques (y compris méningite virale);
- infections oculaires;
- infections bactériennes;
- diverticulite (inflammation et infection du gros intestin);
- tumeurs;
- tumeurs du système lymphatique;
- mélanome;
- troubles du système immunitaire pouvant affecter les poumons, la peau et les ganglions lymphatiques (présentant le plus souvent une sarcoïdose);
- vascularite (inflammation des vaisseaux sanguins);
- tremblement;
- accident vasculaire cérébral;
- neuropathie;
- vision double;
- perte auditive, sonnerie;
- sensation de battements cardiaques irréguliers tels que des palpitations ;
- problèmes cardiaques pouvant provoquer un essoufflement ou un gonflement des chevilles;
- infarctus aigu du myocarde;
- formation d'un sac dans la paroi d'une artère principale, inflammation et caillot dans une veine, obstruction d'un vaisseau sanguin ;
- maladie pulmonaire provoquant un essoufflement (y compris une inflammation);
- embolie pulmonaire (occlusion d'une artère pulmonaire);
- épanchement pleural (collecte anormale de liquide dans l'espace pleural);
- inflammation du pancréas qui provoque une douleur intense dans l'abdomen et le dos;
- difficulté à avaler;
- œdème facial;
- inflammation de la vésicule biliaire, calculs de la vésicule biliaire;
- foie gras;
- sueurs nocturnes;
- cicatrice;
- catabolisme musculaire anormal;
- lupus érythémateux disséminé (y compris inflammation de la peau, du cœur, des poumons, des articulations et d'autres organes)
- sommeil interrompu;
- impuissance;
- inflammatoires.
Rare (peut affecter jusqu'à 1 personne sur 1 000) :
- leucémie (néoplasme malin affectant le système hématopoïétique au niveau périphérique (sang) et la moelle osseuse);
- réaction allergique sévère avec choc;
- sclérose en plaque;
- troubles neurologiques (tels que inflammation du nerf optique et syndrome de Guillain-Barré pouvant entraîner une faiblesse musculaire, des sensations anormales, des picotements dans les bras et le haut du corps) ;
- crise cardiaque;
- fibrose pulmonaire (cicatrisation du poumon);
- perforation intestinale;
- hépatite;
- réactivation de l'hépatite B;
- hépatite auto-immune (inflammation du foie causée par votre propre système immunitaire);
- vascularite cutanée (inflammation des vaisseaux sanguins de la peau) ;
- Syndrome de Stevens-Johnson (les premiers symptômes comprennent un malaise, de la fièvre, des maux de tête et des éruptions cutanées);
- œdème facial associé à des réactions allergiques;
- érythème polymorphe (éruption cutanée inflammatoire);
- syndrome de type lupus.
Fréquence indéterminée (la fréquence ne peut être estimée à partir des données disponibles) :
- lymphome à cellules T hépato-splénique (un cancer du sang rare qui est souvent mortel);
- Carcinome à cellules de Merkel (un type de cancer de la peau);
- Insuffisance hépatique;
- aggravation d'une affection appelée dermatomyosite (se manifestant par une éruption cutanée accompagnée d'une faiblesse musculaire).
Certains des effets indésirables observés avec Humira peuvent être asymptomatiques et ne peuvent être trouvés que dans des analyses de sang. Ceux-ci inclus:
Très fréquent (peut affecter plus de 1 personne sur 10) :
- faible nombre de globules blancs;
- faible nombre de globules rouges;
- augmentation des lipides sanguins;
- augmentation des enzymes hépatiques.
Fréquent (peut affecter jusqu'à 1 personne sur 10) :
- augmentation du nombre de globules blancs;
- numération plaquettaire réduite;
- augmentation de l'acide urique dans le sang;
- altération du sodium dans le sang;
- réduction du calcium dans le sang;
- réduction du phosphore dans le sang;
- augmentation de la glycémie;
- augmentation de la lactate déshydrogénase sanguine;
- présence d'auto-anticorps dans le sang.
Rare (peut affecter jusqu'à 1 personne sur 1 000) :
- faible nombre de globules blancs, de globules rouges et de plaquettes.
Fréquence indéterminée (la fréquence ne peut être estimée à partir des données disponibles) :
- insuffisance hépatique.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien, y compris tout effet indésirable éventuel non mentionné dans cette notice.
Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration figurant à l'annexe V. En déclarant les effets indésirables, vous contribuez à fournir plus d'informations sur la sécurité de ce médicament.
Expiration et conservation
Gardez ce médicament hors de la vue et de la portée des enfants.
N'utilisez pas ce médicament après la date de péremption indiquée sur l'étiquette/le blister/la boîte après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver au réfrigérateur (2°C - 8°C). Ne pas congeler.
Conservez la seringue dans l'emballage approprié pour protéger le médicament de la lumière.
Conditions de stockage alternatives :
En cas de besoin (par exemple en voyage) une seule seringue prête à l'emploi peut être conservée à température ambiante (jusqu'à 25 °C) jusqu'à 14 jours - veillez à protéger le médicament de la lumière. être conservée à température ambiante, la seringue doit être utilisée dans les 14 jours ou jetée, même si elle est remise au réfrigérateur.
Vous devez noter la date à laquelle la seringue est sortie pour la première fois du réfrigérateur et la date après laquelle la seringue doit être jetée.
Ne jetez aucun médicament au tout-à-l'égout ou avec les ordures ménagères.Demandez à votre médecin ou à votre pharmacien comment jeter les médicaments que vous n'utilisez plus.Cela contribuera à protéger l'environnement.
Composition et forme pharmaceutique
Ce que contient Humira
L'ingrédient actif est l'adalimumab.
Les autres composants sont le mannitol, l'acide citrique monohydraté, le citrate de sodium, le dihydrogénophosphate de sodium dihydraté, le phosphate disodique dihydraté, le chlorure de sodium, le polysorbate 80, l'hydroxyde de sodium et l'eau pour préparations injectables.
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose de 0,8 ml, il est donc essentiellement « sans sodium » et ne contient aucun conservateur.
A quoi ressemble la seringue préremplie Humira et contenu de l'emballage extérieur
La seringue préremplie Humira se compose d'une solution d'adalimumab contenue dans une seringue en verre avec un dispositif de sécurité pour aiguille. Chaque boîte contient 1 seringue préremplie avec un dispositif de sécurité pour aiguille à usage hospitalier ou pour administration par un tiers, avec 1 tampon d'alcool.
Humira 40 mg solution injectable en seringues préremplies se présente sous la forme d'une solution stérile de 40 mg d'adalimumab dissous dans 0,8 ml de solution.
Toutes les présentations peuvent ne pas être commercialisées.
Humira est disponible en flacon, en seringue préremplie et en stylo prérempli.
+ Notice d'emballage source : AIFA (Agence italienne des médicaments). Contenu publié en janvier 2016. Les informations présentes peuvent ne pas être à jour.
Pour avoir accès à la version la plus récente, il est conseillé d'accéder au site Internet de l'AIFA (Agence Italienne du Médicament). Avis de non-responsabilité et informations utiles.
01.0 DÉNOMINATION DU MÉDICAMENT
HUMIRA 400 MG SOLUTION POUR INJECTION EN STYLO PRÉREMPLI
02.0 COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque stylo prérempli unidose de 0,8 ml contient 40 mg d'adalimumab.
L'adalimumab est un anticorps monoclonal humain recombinant exprimé dans des cellules d'ovaire de hamster chinois.
Pour la liste complète des excipients, voir rubrique 6.1.
03.0 FORME PHARMACEUTIQUE
Solution injectable limpide contenue dans un stylo prérempli.
04.0 INFORMATIONS CLINIQUES
04.1 Indications thérapeutiques
La polyarthrite rhumatoïde
Humira, en association avec le méthotrexate, est indiqué pour :
§ Traitement des patients adultes atteints de polyarthrite rhumatoïde active modérée à sévère lorsque la réponse aux antirhumatismaux modificateurs de la maladie (ARMM), y compris le méthotrexate, est inadéquate.
§ le traitement de la polyarthrite rhumatoïde sévère, active et évolutive chez l'adulte non préalablement traité par méthotrexate.
Humira peut être administré en monothérapie en cas d'intolérance au méthotrexate ou lorsque la poursuite du traitement par méthotrexate est inappropriée.
Humira, en association avec le méthotrexate, inhibe la progression des lésions structurelles évaluées par radiographie et améliore la fonction physique chez cette population de patients.
Arthrite idiopathique juvénile
Arthrite juvénile idiopathique polyarticulaire
Humira en association avec le méthotrexate est indiqué dans le traitement de l'arthrite juvénile idiopathique polyarticulaire active chez les patients à partir de 2 ans qui ont eu une réponse inadéquate à un ou plusieurs médicaments antirhumatismaux de fond (ARMM).Humira peut être administré en monothérapie. en cas d'intolérance au méthotrexate ou lorsque la poursuite du traitement par méthotrexate est inappropriée (pour l'efficacité en monothérapie, voir rubrique 5.1). Humira n'a pas été étudié chez les patients de moins de 2 ans.
Arthrite associée à une enthésite
Humira est indiqué dans le traitement de l'arthrite active associée à une enthésite chez les patients âgés de 6 ans et plus qui ont eu une réponse inadéquate ou qui sont intolérants au traitement conventionnel (voir rubrique 5.1).
Spondylarthrite axiale
Spondylarthrite ankylosante (SA)
Humira est indiqué pour le traitement des patients adultes atteints de spondylarthrite ankylosante active sévère dont la réponse au traitement conventionnel a été inadéquate.
Spondylarthrite axiale sans signe radiographique de SA
Humira est indiqué pour le traitement des patients adultes atteints de spondylarthrite axiale sévère sans signe radiographique de SA mais avec des signes objectifs d'inflammation détectés par une élévation de la protéine C réactive et/ou une IRM, qui ont eu une réponse inadéquate ou sont intolérants aux médicaments non stéroïdiens. médicaments anti-inflammatoires.
Arthrite psoriasique
Humira est indiqué dans le traitement du rhumatisme psoriasique actif et évolutif chez l'adulte lorsque la réponse aux précédents médicaments antirhumatismaux modificateurs de la maladie (ARMM) a été inadéquate. Humira s'est avéré inadéquat. réduit le taux de progression de l'articulation périphérique associée dommages détectés par les radiographies chez les patients présentant des sous-groupes polyarticulaires symétriques de la maladie (voir rubrique 5.1) et améliore la fonction physique.
Psoriasis
Humira est indiqué dans le traitement du psoriasis en plaques chronique modéré à sévère chez les patients adultes qui n'ont pas répondu, ou qui ont des contre-indications, ou qui ont été intolérants à d'autres thérapies systémiques, y compris le traitement par cyclosporine, méthotrexate ou PUVA.
la maladie de Crohn
Humira est indiqué dans le traitement de la maladie de Crohn active modérée à sévère chez les patients adultes n'ayant pas répondu à une cure complète et adéquate de corticoïdes et/ou d'un immunosuppresseur, ou chez les patients qui sont intolérants à ces traitements ou qui ont des contre-indications médicales à eux.
La maladie de Crohn chez les patients pédiatriques
Humira est indiqué dans le traitement de la maladie de Crohn active sévère chez les patients pédiatriques (à partir de 6 ans) qui ont eu une réponse inadéquate au traitement conventionnel ≤ y compris une thérapie nutritionnelle primaire, une corticothérapie et un immunomodulateur, ou qui sont intolérants ou ont des contre-indications à de telles thérapies.
Rectocolite hémorragique
Humira est indiqué dans le traitement de la rectocolite hémorragique active modérée à sévère chez les patients adultes qui ont eu une réponse inadéquate au traitement conventionnel, y compris les corticoïdes et la 6-mercaptopurine (6-MP) ou l'azathioprine (AZA) ou qui sont intolérants ou ont des contre-indications à ces thérapies.
04.2 Posologie et mode d'administration
Dosage
Le traitement par Humira doit être instauré et surveillé par des médecins spécialistes expérimentés dans le diagnostic et le traitement des affections pour lesquelles Humira est indiqué. Les patients traités par Humira doivent recevoir une carte d'alerte spéciale.
Après avoir reçu des instructions appropriées sur la technique d'injection d'Humira, les patients peuvent s'injecter eux-mêmes si leur médecin le juge approprié, et avec des examens médicaux périodiques si nécessaire.
Pendant le traitement par Humira, les autres thérapies concomitantes (par exemple, corticostéroïdes et/ou agents immunomodulateurs) doivent être optimisées.
La polyarthrite rhumatoïde
La dose d'Humira indiquée chez les patients adultes atteints de polyarthrite rhumatoïde est de 40 mg d'adalimumab en une seule injection sous-cutanée toutes les deux semaines. Le méthotrexate doit être poursuivi pendant le traitement par Humira.
Les glucocorticoïdes, les salicylates, les anti-inflammatoires non stéroïdiens ou les analgésiques peuvent être poursuivis pendant le traitement par Humira. Concernant l'association avec d'autres DMARD autres que le méthotrexate, voir rubriques 4.4 et 5.1.
Certains patients qui présentent une diminution de la réponse en monothérapie peuvent bénéficier d'une augmentation de leur dose à 40 mg d'adalimumab par semaine.
Arrêt de la dose
Il peut être nécessaire d'interrompre l'administration, par exemple avant une intervention chirurgicale ou en cas d'infection sévère.
Les données disponibles indiquent que la réintroduction d'Humira après un arrêt de 70 jours ou plus entraîne une réponse clinique de même importance et avec un profil de sécurité similaire à celui d'avant l'arrêt du traitement.
Spondylarthrite ankylosante, spondylarthrite axiale sans signe radiographique de SA et rhumatisme psoriasique
La dose recommandée d'Humira pour les patients atteints de spondylarthrite ankylosante, de spondylarthrite axiale sans signe radiographique de SA et pour les patients atteints de rhumatisme psoriasique est de 40 mg d'adalimumab administrés toutes les deux semaines en une dose unique par voie sous-cutanée.
Pour toutes les indications ci-dessus, les données disponibles suggèrent que la réponse clinique est généralement obtenue dans les 12 semaines suivant le début du traitement. En cas d'absence de réponse au cours de cette période, la poursuite du traitement doit être soigneusement envisagée.
Psoriasis
La dose recommandée d'Humira pour les patients adultes est une dose initiale de 80 mg, administrée par voie sous-cutanée, suivie d'une dose de 40 mg, par voie sous-cutanée, administrée toutes les deux semaines, en commençant la semaine suivant la " prise de la dose initiale.
Une attention particulière doit être accordée à la poursuite du traitement au-delà de 16 semaines si les patients n'ont pas développé de réponse satisfaisante au cours de cette période.
la maladie de Crohn
La dose indiquée d'Humira pour le traitement d'induction est de 80 mg à la semaine 0 pour les patients adultes atteints de la maladie de Crohn modérée à sévère, suivie de 40 mg à la semaine 2. Si une réponse plus rapide au traitement est nécessaire, une dose de 160 mg à la semaine 0 peut (cette dose peut être administrée sous forme de quatre injections sur une journée ou de deux injections par jour pendant deux jours consécutifs), suivies de 80 mg à la semaine 2, sachant que le risque d'événements indésirables est plus important lors de l'induction.
Après le traitement d'induction, la dose indiquée est de 40 mg toutes les deux semaines, administrée par voie sous-cutanée. Alternativement, si un patient a arrêté le traitement par Humira et que les symptômes de la maladie réapparaissent, le traitement par Humira peut être ré-administré. Il existe peu de données sur la ré-administration d'Humira si une période de 8 semaines s'est écoulée depuis l'administration de la dose précédente.
Pendant le traitement d'entretien, la posologie des corticostéroïdes peut être progressivement réduite selon les directives élaborées pour la prise en charge clinique de la maladie.
Certains patients dont la réponse au traitement est réduite peuvent bénéficier d'une augmentation de la fréquence d'administration à 40 mg d'Humira par semaine.
Les patients qui n'ont pas montré de réponse adéquate au traitement à la semaine 4 peuvent bénéficier de la mise en place d'un traitement d'entretien continu jusqu'à la semaine 12. Chez les patients dont la réponse au traitement est inadéquate au cours de cette période, il convient d'évaluer soigneusement la nécessité de poursuivre le traitement.
Rectocolite hémorragique
Le schéma posologique d'induction recommandé d'Humira pour les patients adultes atteints de rectocolite hémorragique modérée à sévère est de 160 mg à la semaine 0 (la dose peut être administrée en 4 injections en une journée ou en deux injections par jour, pendant deux jours consécutifs) et 80 mg par semaine 2. Après le traitement d'induction, la dose recommandée est de 40 mg toutes les deux semaines par voie sous-cutanée.
Pendant le traitement d'entretien, les corticoïdes peuvent être progressivement réduits selon les recommandations de pratique clinique.
Les patients présentant une réponse réduite documentée peuvent bénéficier d'une augmentation de la fréquence d'administration à 40 mg d'Humira par semaine.
Les données disponibles suggèrent que la réponse clinique est généralement obtenue dans les 2 à 8 semaines suivant le traitement.
Le traitement par Humira ne doit pas être poursuivi chez les patients qui n'ont pas répondu pendant cette période.
Les personnes plus âgées
Aucun changement de dosage n'est nécessaire.
Insuffisance hépatique et/ou rénale
Humira n'a pas été étudié dans ces populations de patients. Aucune recommandation posologique ne peut être donnée.
Population pédiatrique
Arthrite idiopathique juvénile
Arthrite juvénile idiopathique polyarticulaire de 2 à 12 ans
La dose recommandée d'Humira pour les patients atteints d'arthrite juvénile idiopathique polyarticulaire âgés de 2 à 12 ans est de 24 mg/m2 de surface corporelle jusqu'à une dose unique maximale de 20 mg d'adalimumab (pour les patients âgés de 2 ans - taille et poids du patient ( Tableau 1) Un flacon pédiatrique de 40 mg est disponible pour les patients qui doivent prendre une dose inférieure à la dose maximale de 40 mg.
Tableau 1. Dose d'Humira en millilitres (ml) en fonction de la taille et du poids chez les patients atteints d'arthrite juvénile idiopathique polyarticulaire et d'arthrite associée à l'enthésite
* La dose unique maximale est de 40 mg (0,8 ml)
Arthrite juvénile idiopathique polyarticulaire à partir de 13 ans
Pour les patients de 13 ans et plus, une dose de 40 mg est administrée toutes les deux semaines, quelle que soit la surface corporelle.
Les données disponibles suggèrent que la réponse clinique est généralement obtenue dans les 12 semaines suivant le traitement. Chez les patients dont la réponse au traitement est inadéquate au cours de cette période, la nécessité de poursuivre le traitement doit être soigneusement étudiée.
Il n'y a pas d'utilisation justifiée d'Humira chez les patients de moins de 2 ans dans cette indication.
Arthrite associée à une enthésite
La dose recommandée d'Humira chez les patients atteints d'arthrite associée à l'enthésite, âgés de 6 ans et plus, est de 24 mg/m2 de surface corporelle, jusqu'à une dose unique maximale de 40 mg d'adalimumab administrée toutes les deux semaines par injection sous-cutanée. Le volume d'injection est choisi en fonction de la taille et du poids du patient (tableau 1).
Humira n'a pas été étudié chez les patients de moins de 6 ans atteints d'arthrite associée à l'enthésite.
Psoriasis pédiatrique
La sécurité et l'efficacité d'Humira chez les enfants âgés de 4 à 17 ans n'ont pas été établies. Aucune donnée n'est disponible. Il n'y a pas d'utilisation justifiée d'Humira chez les enfants âgés de moins de 4 ans dans cette indication.
La maladie de Crohn chez les patients pédiatriques
La maladie de Crohn chez les patients pédiatriques
La dose d'induction recommandée d'Humira chez les sujets pédiatriques atteints d'une maladie de Crohn sévère est de 40 mg à la semaine 0 suivi de 20 mg à la semaine 2. Si une réponse plus rapide au traitement est requise, un schéma posologique de 80 mg à la semaine 0 peut être utilisé ( dose peut être administrée en deux injections en une journée) et 40 mg à la semaine 2, étant entendu que le risque d'événements indésirables peut être plus élevé avec l'utilisation de la dose d'induction plus élevée.
Après le traitement d'induction, la dose recommandée est de 20 mg toutes les deux semaines en injection sous-cutanée. Certaines personnes présentant une réponse insuffisante peuvent bénéficier d'une augmentation de la fréquence des doses d'Humira 20 mg chaque semaine.
Maladie de Crohn chez les patients pédiatriques ≥ 40 kg :
La dose d'induction recommandée d'Humira chez les sujets pédiatriques atteints d'une maladie de Crohn sévère est de 80 mg à la semaine 0 suivi de 40 mg à la semaine 2. Si une réponse plus rapide au traitement est requise, un schéma posologique de 160 mg à la semaine 0 peut être utilisé ( la dose peut être administrée en quatre injections par jour ou en deux injections par jour pendant deux jours consécutifs), et 80 mg à la semaine 2, étant entendu que le risque d'événements indésirables peut être plus élevé avec l'utilisation de la dose de induction.
Après le traitement d'induction, la dose recommandée est de 40 mg toutes les deux semaines en injection sous-cutanée. Certaines personnes présentant une réponse insuffisante peuvent bénéficier d'une augmentation de la fréquence des doses à 40 mg d'Humira par semaine.
La poursuite du traitement doit être soigneusement envisagée chez un sujet ne répondant pas à la semaine 12.
Il n'y a pas d'utilisation justifiée d'Humira chez les enfants de 6 ans et moins dans cette indication.
Colite ulcéreuse pédiatrique
La sécurité et l'efficacité d'Humira chez les enfants âgés de 4 à 17 ans n'ont pas encore été établies. Aucune donnée n'est disponible. Il n'y a pas d'utilisation justifiée d'Humira chez les enfants âgés de moins de 4 ans dans cette indication.
Arthrite psoriasique et spondylarthrite axiale y compris spondylarthrite ankylosante
Il n'y a pas d'utilisation justifiée d'Humira dans la population pédiatrique dans les indications spondylarthrite ankylosante et rhumatisme psoriasique.
Mode d'administration
Humira est administré par injection sous la peau. Des instructions complètes d'utilisation sont fournies dans la notice.
Un flacon pédiatrique de 40 mg est disponible pour les patients nécessitant une administration inférieure à la dose complète de 40 mg.
04.3 Contre-indications
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique 6.1.
Tuberculose active ou autres infections graves telles que sepsis et infections opportunistes (voir rubrique 4.4).
Insuffisance cardiaque modérée à sévère (NYHA classe III/IV) (voir rubrique 4.4).
04.4 Mises en garde spéciales et précautions d'emploi appropriées
Afin d'améliorer la traçabilité des médicaments biologiques, la marque et le numéro de lot du produit administré doivent être clairement enregistrés (ou marqués).
Infections
Les patients traités par anti-TNF sont plus sensibles aux infections sévères. Une fonction pulmonaire altérée peut augmenter le risque de développer des infections.
Les patients doivent donc être soigneusement dépistés pour les infections, y compris la tuberculose, avant, pendant et après le traitement par Humira. L'élimination de l'adalimumab pouvant prendre jusqu'à quatre mois, la surveillance doit être poursuivie pendant cette période.
Le traitement par Humira ne doit pas être instauré chez les patients présentant des infections actives, y compris des infections chroniques ou localisées, tant que celles-ci ne sont pas maîtrisées. Chez les patients qui ont été exposés à la tuberculose et chez les patients qui ont voyagé dans des zones à haut risque de tuberculose ou de mycose endémique, telles que l'histoplasmose, la coccidioïdomycose ou la blastomycose, le risque et le bénéfice du traitement par Humira doivent être pris en compte avant d'initier le traitement (voir Infections opportunistes).
Les patients qui développent une nouvelle infection pendant le traitement par Humira doivent être suivis de près et subir une évaluation diagnostique complète. En cas d'apparition d'une nouvelle infection grave ou d'un sepsis, l'administration d'Humira doit être interrompue et un traitement antimicrobien ou antifongique approprié instauré jusqu'à ce que l'infection soit contrôlée. prédisposent les patients aux infections, y compris l'utilisation concomitante de médicaments immunosuppresseurs.
Infections graves:
Des cas d'infections graves, notamment de septicémie, causées par des bactéries, des mycobactéries, des champignons invasifs, des parasites, des virus ou d'autres infections opportunistes telles que la listériose, la légionellose et la pneumocystose ont été rapportés chez des patients traités par Humira.
Les autres infections graves observées dans les essais cliniques comprennent la pneumonie, la pyélonéphrite, l'arthrite septique et la septicémie. Des cas d'hospitalisation ou d'événements mortels associés à des infections ont été rapportés.
Tuberculose:
Des cas de tuberculose, y compris une réactivation et une nouvelle apparition de tuberculose, ont été signalés chez des patients utilisant Humira. Des cas de tuberculose pulmonaire et extrapulmonaire (c'est-à-dire disséminée) ont été rapportés.
Avant de commencer le traitement par Humira, tous les patients doivent être examinés pour rechercher la présence d'une tuberculose active ou inactive (« latente »). Cette évaluation doit inclure « les antécédents médicaux détaillés des patients ayant des antécédents de tuberculose ou tout contact avec des personnes atteintes de tuberculose active, et ayant reçu des traitements immunosuppresseurs antérieurs et/ou concomitants. ) chez tous les patients (les directives locales peuvent être suivies). Il est recommandé d'effectuer ces tests et d'enregistrer les résultats sur la carte d'alerte du patient. Les médecins doivent être attentifs au risque de résultats faussement négatifs au test cutané à la tuberculine, en particulier chez les patients gravement malades ou immunodéprimés.
Si une tuberculose active est diagnostiquée, le traitement par Humira ne doit pas être instauré (voir rubrique 4.3).
Dans toutes les situations décrites ci-dessous, une « évaluation minutieuse du rapport risque/bénéfice du traitement par Humira doit être effectuée.
En cas de suspicion de tuberculose latente, il est conseillé de consulter un médecin spécialisé dans le traitement de la tuberculose.
Si une tuberculose latente est diagnostiquée, un traitement prophylactique antituberculeux doit être instauré conformément aux recommandations locales avant d'initier le traitement par Humira.
La mise en place d'un traitement prophylactique antituberculeux doit également être envisagée avant de commencer le traitement par Humira chez les patients présentant des facteurs de risque de tuberculose différents ou significatifs malgré un test de tuberculose négatif et chez les patients dont les antécédents médicaux présentent des antécédents personnels de tuberculose latente ou active en dont il n'est pas possible de confirmer si le traitement qu'ils ont suivi était adéquat.
Malgré le traitement prophylactique de la tuberculose, une réactivation de la tuberculose est survenue chez des patients traités par Humira. Certains patients traités avec succès pour une tuberculose active ont de nouveau souffert de tuberculose pendant le traitement par Humira.
Il doit être conseillé aux patients de consulter un médecin si des signes/symptômes évoquant une éventuelle infection tuberculeuse (par exemple toux persistante, amaigrissement, perte de poids, fièvre modérée, apathie) surviennent pendant ou après le traitement par Humira.
Autres infections opportunistes:
Des cas d'infections opportunistes, y compris des infections fongiques invasives, ont été observés chez des patients prenant Humira. Ces infections n'ont pas été correctement diagnostiquées chez les patients prenant des anti-TNF et cela a entraîné un retard dans le traitement approprié, parfois avec une issue fatale.
Chez les patients qui développent des signes et symptômes tels que fièvre, malaise, perte de poids, sueurs, toux, dyspnée et/ou infiltrat pulmonaire ou autre maladie systémique grave avec ou sans choc concomitant, une infection fongique invasive doit être suspectée et arrêtée rapidement. Humira Le diagnostic et l'administration d'un traitement antifongique empirique chez ces patients doivent être effectués en consultation avec un médecin spécialisé dans le traitement des patients atteints d'infections fongiques invasives.
Réactivation de l'hépatite B
Une réactivation de l'hépatite B (par exemple, antigène de surface positif) s'est produite chez des porteurs chroniques du virus de l'hépatite B traités par des antagonistes du TNF, y compris Humira. Certains cas ont eu une issue fatale. Avant de commencer le traitement par Humira, les patients doivent être testés pour l'infection par le virus de l'hépatite B. La consultation d'un médecin expérimenté dans le traitement de l'hépatite B est recommandée pour les patients dont le test est positif pour le virus de l'hépatite B. hépatite B.
Les porteurs du virus de l'hépatite B nécessitant un traitement par Humira doivent être étroitement surveillés afin de détecter tout signe et symptôme d'infection active par le virus de l'hépatite B, non seulement tout au long du traitement, mais également pendant les mois suivant l'arrêt du traitement. avec le virus de l'hépatite B sous traitement antiviral afin d'éviter la réactivation du virus de l'hépatite B en même temps qu'un traitement par anti-TNF. Chez les patients qui développent une réactivation du virus de l'hépatite B, l'administration d'Humira doit être interrompue et un traitement antiviral efficace doit être instauré accompagné un traitement de soutien adéquat.
Événements neurologiques
Les anti-TNF, y compris Humira, ont été associés dans de rares cas à l'apparition ou à l'exacerbation de symptômes cliniques et/ou de signes radiographiques de maladies démyélinisantes du système nerveux central, y compris la sclérose en plaques, la névrite optique et les maladies démyélinisantes périphériques, y compris le syndrome de Guillain-Barré. Des précautions doivent être prises lors de l'utilisation d'Humira chez les patients présentant des troubles démyélinisants du système nerveux central ou périphérique antérieurs ou récents.
Réactions allergiques
Dans les essais cliniques, les réactions allergiques graves associées à Humira ont été rares. Les réactions allergiques non graves associées à Humira au cours des essais cliniques ont été peu fréquentes. Des cas de réactions allergiques graves, notamment d'anaphylaxie, ont été rapportés après l'administration d'Humira. En cas de survenue de réactions anaphylactiques ou d'autres manifestations allergiques sévères, l'administration d'Humira doit être interrompue immédiatement et un traitement approprié initié.
Immunosuppression
Dans une étude portant sur 64 patients atteints de polyarthrite rhumatoïde, recevant un traitement par Humira, aucun signe d'inhibition de l'hypersensibilité retardée, de réduction des taux d'immunoglobulines ou de modification du nombre de lymphocytes T, B, NK, monocytes/cellules, macrophages et neutrophiles n'a été mis en évidence.
Tumeurs et maladies lymphoprolifératives
Dans les sections contrôlées des essais cliniques sur les antagonistes du TNF, plus de cas de tumeurs malignes, y compris de lymphomes, ont été observés chez les patients recevant des anti-TNF que dans le groupe témoin. Cependant, les cas étaient rares. Dans les études post-commercialisation, des cas de leucémie ont été rapportés chez des patients traités par un anti-TNF. Il existe un risque accru de développer des lymphomes et des leucémies chez les patients atteints de polyarthrite rhumatoïde sévèrement active et de longue durée, une maladie inflammatoire qui complique l'évaluation du risque.Dans l'état actuel des connaissances, le développement de lymphomes ne peut être exclu. traités avec des anti-TNF.
Des cas de cancers, certains mortels, ont été rapportés chez des enfants, des adolescents et de jeunes adultes (jusqu'à 22 ans) traités par des anti-TNF (initiation du traitement ≤ 18 ans), dont l'adalimumab dans les études post-commercialisation. Environ la moitié des cas étaient des lymphomes. Les autres cas représentaient une multiplicité de cancers différents et comprenaient des cancers rares généralement associés à une immunosuppression. Un risque de développement de tumeurs chez les enfants et adolescents traités par anti-TNF ne peut être exclu.
De rares cas de lymphome hépatosplénique à cellules T ont été observés après commercialisation chez des patients traités par adalimumab. Ce type rare de lymphome à cellules T a une évolution clinique très agressive et est souvent fatale. Certains de ces cas de lymphome hépatosplénique à cellules T sont survenus chez de jeunes patients adultes traités par Humira et recevant un traitement concomitant par azathioprine ou 6-mercaptopurine, médicaments utilisés pour traiter les maladies inflammatoires de l'intestin. Le risque potentiel de l'association d'azathioprine ou de 6-mercaptopurine et d'Humira doit être soigneusement évalué. Un risque de développer un lymphome T hépatosplénique ne peut être exclu chez les patients traités par Humira (voir rubrique 4.8).
Aucune étude clinique n'a été menée chez des patients ayant des antécédents de cancer ou chez des patients dont le traitement par Humira s'est poursuivi après le développement d'un cancer. Par conséquent, le traitement par Humira dans cette population de patients doit être envisagé avec une prudence supplémentaire (voir rubrique 4.8).
Avant et pendant le traitement par Humira, tous les patients, en particulier ceux ayant des antécédents de traitement immunosuppresseur intensif ou ceux atteints de psoriasis ayant des antécédents de traitement par PUVA, doivent être examinés pour rechercher la présence d'un éventuel cancer cutané non mélanique. Des mélanomes et des carcinomes à cellules de Merkel ont également été rapportés chez des patients traités par des anti-TNF, dont l'adalimumab (voir rubrique 4.8).
Dans un essai clinique exploratoire évaluant l'utilisation d'un autre antagoniste du TNF, l'infliximab, chez des patients atteints de bronchopneumopathie chronique obstructive (BPCO) modérée à sévère, plus de tumeurs malignes ont été rapportées chez les patients traités par infliximab que chez les patients témoins, en particulier dans les poumons ou la tête. et cou Tous les patients avaient des antécédents de gros fumeurs.
Par conséquent, il convient d'être prudent lors de l'utilisation de tout antagoniste du TNF chez les patients atteints de BPCO, ainsi que chez les patients présentant un risque accru de malignité en raison d'un tabagisme excessif.
Sur la base des données actuelles, on ne sait pas si le traitement par l'adalimumab affecte le risque de développer une dysplasie ou un cancer du côlon. Tous les patients atteints de rectocolite hémorragique qui présentent un risque accru de dysplasie ou de carcinome du côlon (par exemple, les patients atteints de rectocolite hémorragique de longue date ou de cholangite sclérosante primitive), ou qui ont eu des antécédents de dysplasie ou de cancer du côlon doivent être dépistés régulièrement pour la dysplasie tout au long de la maladie. Cette évaluation doit inclure des coloscopies et des biopsies basées sur les recommandations locales.
Réactions affectant le système hématopoïétique
De rares cas de pancytopénie, y compris la survenue d'anémie aplasique, ont été rapportés suite à l'utilisation d'antagonistes du TNF.Evénements indésirables affectant le système hématopoïétique, dont des cytopénies importantes car d'un point de vue médical (par exemple, thrombocytopénie, leucopénie) Au cours traitement par Humira tous les patients doivent être informés de la nécessité de consulter immédiatement un médecin afin d'obtenir une assistance adéquate si des signes et symptômes évocateurs d'une dyscrasie se développent (par exemple fièvre persistante, ecchymoses, hémorragie, pâleur). système hématopoïétique, la nécessité d'arrêter le traitement par Humira doit être envisagée.
Vaccination
Des réponses en anticorps similaires au vaccin standard contre le pneumocoque 23-valent et au vaccin trivalent contre le virus de la grippe ont été observées dans une étude portant sur 226 sujets adultes atteints de polyarthrite rhumatoïde qui ont été traités par adalimumab ou un placebo. patients prenant Humira.
Chez les patients pédiatriques, il est recommandé que le calendrier de vaccination prévu soit mis en œuvre, si possible, conformément aux directives de vaccination en vigueur avant d'initier un traitement à base d'Humira.
Les patients traités par Humira peuvent recevoir des vaccinations simultanées, à l'exception des vaccins vivants. L'administration de vaccins vivants aux nourrissons exposés à l'adalimumab in utero n'est pas recommandée dans les 5 mois suivant la dernière administration d'adalimumab par la mère pendant la grossesse.
Insuffisance cardiaque congestive
Une aggravation de l'insuffisance cardiaque congestive et une mortalité accrue associée ont été observées dans un essai clinique avec un autre médicament anti-TNF. Une aggravation de l'insuffisance cardiaque congestive a également été observée chez les patients traités par Humira. Humira doit être utilisé avec prudence chez les patients présentant une insuffisance cardiaque légère (NYHA classe I/II). Humira est contre-indiqué en cas d'insuffisance cardiaque modérée ou sévère (voir rubrique 4.3).Le traitement par Humira doit être interrompu chez les patients présentant une aggravation ou de nouveaux symptômes d'insuffisance cardiaque congestive.
Processus auto-immuns
Le traitement par Humira peut induire la formation d'anticorps auto-immuns. L'impact d'un traitement à long terme par Humira sur le développement de maladies auto-immunes n'est pas connu. Si un patient développe des symptômes évocateurs d'un syndrome de type lupus après un traitement par Humira et est positif pour les anticorps anti-ADN double brin, ne poursuivez pas le traitement par Humira. doit être administré (voir rubrique 4.8).
Administration concomitante d'antagonistes du DMARDS ou du TNF biologiques
Des infections graves sans bénéfice clinique par rapport à l'étanercept seul ont été observées dans les essais cliniques sur l'association de l'anakinra et d'un autre anti-TNF, l'étanercept.
Compte tenu du type d'événements indésirables observés avec l'association d'anakinra et d'étanercept, des effets indésirables similaires peuvent survenir suite à l'association d'anakinra et d'un autre anti-TNF. Par conséquent, l'association de l'adalimumab et de l'anakinra n'est pas recommandée (voir rubrique 4.5).
L'administration concomitante d'adalimumab avec d'autres DMARD biologiques (par exemple anakinra et abatacept) ou d'autres antagonistes du TNF n'est pas recommandée en raison d'un risque accru d'infections, y compris d'infections graves et d'autres interactions médicamenteuses potentielles (voir rubrique 4.5).
Interventions chirurgicales
Il existe une expérience « limitée » chez les patients traités par Humira en ce qui concerne la sécurité des interventions chirurgicales. La longue demi-vie de l'adalimumab doit être prise en compte lors de la planification de la chirurgie. Un patient qui subit une intervention chirurgicale tout en étant traité par Humira doit être suivi de près pour le développement d'infections, auquel cas des mesures doivent être prises. Il existe une expérience « limitée » concernant la sécurité chez les patients subissant une chirurgie de remplacement articulaire pendant qu'ils prennent Humira.
Obstruction de l'intestin grêle
L'absence de réponse au traitement de la maladie de Crohn peut indiquer la présence d'une sténose fibreuse rigide qui peut nécessiter une intervention chirurgicale. Les données disponibles suggèrent qu'Humira ne s'aggrave pas et ne provoque pas de sténose.
Les personnes plus âgées
La fréquence des infections graves chez les patients traités par Humira âgés de plus de 65 ans (3,5%) était plus élevée que ceux de moins de 65 ans (1,5%). Certains d'entre eux ont eu une issue fatale. Une attention particulière doit être portée au risque infectieux dans le traitement des patients âgés.
Population pédiatrique
Voir Vaccinations ci-dessus.
04.5 Interactions avec d'autres médicaments et autres formes d'interactions
Le traitement par Humira a été étudié en monothérapie et en association avec le méthotrexate chez des patients atteints de polyarthrite rhumatoïde, d'arthrite juvénile idiopathique polyarticulaire et de rhumatisme psoriasique. La formation d'anticorps était plus faible lorsque Humira était administré en association avec le méthotrexate qu'avec la monothérapie.L'administration d'Humira sans méthotrexate a entraîné une augmentation de la formation d'anticorps, une augmentation de la clairance et une diminution de l'efficacité de l'adalimumab (voir rubrique 5.1).
L'association d'Humira et d'anakinra n'est pas recommandée (voir rubrique 4.4 « Administration concomitante d'ARMM biologiques ou d'antagonistes du TNF »).
L'association d'Humira et de l'abatacept n'est pas recommandée (voir rubrique 4.4 « Administration concomitante d'ARMM biologiques ou d'antagonistes du TNF »).
04.6 Grossesse et allaitement
Grossesse
Pour Humira, des données cliniques limitées sur les grossesses exposées sont disponibles.
Dans une étude de toxicologie du développement menée chez le singe, aucune toxicité maternelle, embryotoxicité ou tératogénicité n'a été trouvée. Aucune donnée préclinique sur la toxicité postnatale de l'adalimumab n'est disponible (voir rubrique 5.3).
En raison de l'inhibition du TNFα, l'administration d'adalimumab pendant la grossesse peut interférer avec la réponse immunitaire normale du nouveau-né. L'administration d'adalimumab n'est donc pas recommandée pendant la grossesse.
L'adalimumab peut traverser le placenta et atteindre le sérum des bébés nés de mères traitées par l'adalimumab pendant la grossesse. Par conséquent, ces enfants sont soumis à un plus grand risque d'infection. L'administration de vaccins vivants aux nourrissons exposés à l'adalimumab in utero n'est pas recommandée dans les 5 mois suivant la dernière administration d'adalimumab par la mère pendant la grossesse.
L'heure du repas
On ne sait pas si l'adalimumab est excrété dans le lait maternel ou absorbé par voie systémique après ingestion.
Cependant, étant donné que les immunoglobulines humaines sont excrétées dans le lait, les femmes ne doivent pas allaiter pendant au moins cinq mois après leur dernier traitement par Humira.
La fertilité
Aucune donnée préclinique sur les effets de l'adalimumab sur la fertilité n'est disponible.
Les femmes en âge de procréer. Contraception chez les hommes et les femmes
Les femmes en âge de procréer doivent utiliser une contraception adéquate pour éviter une grossesse et continuer son utilisation pendant au moins cinq mois après le dernier traitement par Humira.
04.7 Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Humira a des effets mineurs sur l'aptitude à conduire des véhicules ou à utiliser des machines. Des vertiges et des troubles visuels peuvent survenir après l'administration d'Humira (voir rubrique 4.8).
04.8 Effets indésirables
Humira a été étudié chez 8 198 patients dans le cadre d'essais cliniques pivots contrôlés et en ouvert pendant une période allant jusqu'à 60 mois ou plus. Ces études ont été réalisées chez des patients atteints de polyarthrite rhumatoïde d'apparition précoce et de longue durée, d'arthrite juvénile idiopathique (arthrite juvénile idiopathique polyarticulaire et arthrite associée à l'enthésite), ainsi que chez des patients atteints de spondylarthrite axiale (spondylarthrite ankylosante et spondylarthrite axiale sans évidence radiologique). de SA), le rhumatisme psoriasique, la maladie de Crohn, la rectocolite hémorragique et le psoriasis. Les études pivotales contrôlées ont été menées chez 5 343 patients recevant Humira et 3 148 patients recevant un placebo ou un comparateur actif au cours de la période de contrôle.
Le pourcentage de patients ayant arrêté le traitement en raison d'événements indésirables au cours de la phase contrôlée en double aveugle des études pivots était de 6,1 % pour les patients prenant Humira et de 5,7 % pour les patients traités par le groupe témoin.
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés sont les infections (telles que rhinopharyngite, infection des voies respiratoires supérieures et sinusite), les réactions au site d'administration (érythème, prurit, hémorragie, douleur ou gonflement), les céphalées et les douleurs musculo-squelettiques.
Des effets indésirables graves ont été rapportés pour Humira. Les médicaments bloquant le TNF, tels que Humira, affectent le système immunitaire et leur utilisation peut affecter les défenses de l'organisme contre les infections et le cancer.
Des cas d'infections mortelles (y compris des cas de sepsis, d'infections opportunistes et de tuberculose), de réactivation de l'infection par le VHB et de divers types de tumeurs malignes (y compris des cas de leucémie, de lymphome et d'hépato-lymphome) ont également été rapportés après l'administration d'Humira. cellules).
Des réactions hématologiques, neurologiques et auto-immunes sévères ont également été rapportées. Ces derniers comprennent de rares cas de pancytopénie, d'anémie aplasique, d'événements de démyélinisation centrale et périphérique et des cas de lupus, d'affections liées au lupus et de syndrome de Stevens-Johnson.
Population pédiatrique
Effets indésirables chez les patients pédiatriques
En général, les événements indésirables chez les patients pédiatriques étaient similaires à ceux observés chez les patients adultes en termes de fréquence et de type.
Tableau de la liste des effets indésirables
La liste suivante des effets indésirables est basée sur l'expérience des essais cliniques et de l'expérience post-commercialisation et est classée par système/organe impliqué et fréquence (très fréquent ≥1/10 ; fréquent ≥1/100 à
Tableau 2
Effets secondaires
* d'autres informations sont contenues dans les rubriques 4.3, 4.4 et 4.8
** y compris les études d'extension en ouvert
1) y compris les données des déclarations spontanées
Description des effets indésirables sélectionnés
Réactions au site d'injection
Dans les essais cliniques pivots contrôlés chez l'adulte et l'enfant, 13,6 % des patients traités par Humira ont présenté des réactions au site d'injection (érythème et/ou prurit, hémorragie, douleur ou œdème), versus 7,6 % des patients traités par placebo ou contrôle actif. n'a généralement pas nécessité l'arrêt du traitement.
Infections
Dans les essais cliniques pivots contrôlés chez l'adulte et l'enfant, le taux d'infection était de 1,52 par patient/an dans le groupe Humira et de 1,45 par patient/an dans les groupes placebo et contrôle actif.Les infections étaient principalement représentées par les rhinopharyngites, les infections des voies respiratoires supérieures et infection des voies urinaires.La plupart des patients ont continué à prendre Humira après la disparition de l'infection.
L'incidence des infections graves a été de 0,04 par patient/an dans le groupe Humira et de 0,03 par patient/an dans les groupes placebo et contrôle actif.
Dans les études contrôlées et ouvertes avec Humira chez l'adulte et l'enfant, des infections graves (y compris des infections mortelles, qui ne sont survenues que rarement), y compris des cas de tuberculose (y compris des localisations miliaires et extrapulmonaires) ont été rapportées) et des infections opportunistes invasives ( par exemple, de l'histoplasmose disséminée ou extrapulmonaire, la blastomycose, la coccidioïdomycose, la pneumocystose, la candidose, l'aspergillose et la listériose). La plupart des cas de tuberculose sont survenus dans les huit premiers mois suivant le début du traitement et peuvent être interprétés comme une résurgence de la maladie latente.
Tumeurs et maladies lymphoprolifératives
Dans les études réalisées en administrant Humira à des patients atteints d'arthrite juvénile idiopathique (arthrite juvénile idiopathique polyarticulaire et arthrite associée à une enthésite), aucune tumeur maligne n'a été observée chez 249 patients pédiatriques avec une exposition de 655,6 patients-années. patients avec une exposition de 258,9 années-patients au cours d'une étude ayant administré Humira à des patients pédiatriques atteints de la maladie de Crohn.
Dans des sections contrôlées d'études pivotales chez l'adulte avec Humira d'une durée d'au moins 12 semaines chez des patients atteints de polyarthrite rhumatoïde active modérée à sévère, de spondylarthrite ankylosante, de spondylarthrite axiale sans signes radiographiques de SA, de rhumatisme psoriasique, de psoriasis, de maladie de Crohn et de maladie ulcéreuse de colite, de néoplasmes, comme ainsi que le lymphome et le cancer cutané non mélanique, ont été observés à un taux (intervalle de confiance à 95 %) de 6,0 (3,7 ; 9,8) pour 1 000 patients-années chez 4 622 patients traités par Humira versus un taux de 5,1 (2,4, 10,7) par 1 000 patients-années de 2 828 patients témoins (la durée médiane de traitement était de 5,1 mois pour les patients traités par Humira et de 4,0 mois pour les patients témoins). Le taux (intervalle de confiance à 95 %) de cancers cutanés non mélaniques était de 9,7 (6,6 ; 14,3) pour 1 000 patients-années chez les patients traités par Humira et de 5,1 (2,4 ; 10, 7) pour 1 000 patients-années chez les patients témoins. Parmi ces cancers de la peau, les carcinomes épidermoïdes sont survenus à des taux (intervalle de confiance à 95 %) de 2,6 (1,2 ; 5,5) pour 1 000 patients-années chez les patients traités par Humira et de 0,7 (0,1 ; 5,2) pour 1 000 patients-années chez les patients témoins. Le taux (intervalle de confiance à 95 %) de lymphomes était de 0,7 (0,2 à 3,0) pour 1 000 patients-années chez les patients traités par Humira et de 1,5 (0,4 à 5,8) pour 1 000 patients-années chez les patients témoins.
Lorsque des parties de ces études et des études d'extension en ouvert en cours et terminées d'une durée moyenne d'environ 3,4 ans incluant 5 727 patients et plus de 24 568 patients-années de traitement sont combinées, le taux de néoplasmes observés, autres que les lymphomes et les peaux non mélaniques cancer, est d'environ 8,8 pour 1 000 patients-années. Le taux observé de cancer de la peau non mélanique est d'environ 10,3 pour 1 000 patients-années et le taux observé de lymphomes est d'environ 1,4 pour 1 000 patients-années.
Dans une expérience post-commercialisation de janvier 2003 à décembre 2010, principalement chez des patients atteints de polyarthrite rhumatoïde, le taux rapporté de néoplasmes est d'environ 2,7 pour 1 000 traitements/patients-années. Les taux rapportés de cancers cutanés non mélaniques et de lymphomes sont respectivement d'environ 0,2 et 0,3 pour 1 000 traitement/patient-années (voir rubrique 4.4).
De rares cas de lymphome hépatosplénique à cellules T ont été rapportés depuis la commercialisation chez des patients traités par adalimumab (voir rubrique 4.4).
Auto-anticorps
Dans les études IV sur la polyarthrite rhumatoïde, des échantillons de sérum de patients ont été testés à diverses occasions pour rechercher des auto-anticorps. avaient des valeurs positives à la semaine 24. Deux des 3441 patients traités par Humira dans tous les cas de polyarthrite rhumatoïde et de rhumatisme psoriasique ont présenté des signes cliniques indiquant l'apparition d'un syndrome de type lupus. Les patients se sont améliorés après l'arrêt du traitement. Aucun patient n'a développé de néphrite lupique ou centrale symptômes du système nerveux.
Evénements hépato-biliaires
Dans les essais cliniques contrôlés de phase 3 d'Humira chez des patients atteints de polyarthrite rhumatoïde et de rhumatisme psoriasique avec une période de suivi allant de 4 à 104 semaines, des élévations d'ALAT supérieures ou égales à 3 fois la valeur normale maximale sont survenues chez 3,7 % des patients patients traités et 1,6 % des patients témoins.
Dans les essais cliniques contrôlés de phase 3 d'Humira chez des patients atteints de psoriasis en plaques avec une durée de période de contrôle allant de 12 à 24 semaines, des élévations d'ALAT supérieures ou égales à 3 fois la valeur normale maximale sont survenues chez « 1,8 % des patients traités par Humira et 1,8 % des patients traités par Humira. % de patients traités par le contrôle.
Dans les essais cliniques contrôlés de phase 3 d'Humira chez des patients atteints d'arthrite juvénile idiopathique polyarticulaire âgés de 4 à 17 ans et chez des patients atteints d'arthrite associée à une enthésite, âgés de 6 à 17 ans, des élévations d'ALAT supérieures ou égales à un triple de la LSN est survenue chez 6,1 % des patients traités par Humira et 1,3 % des patients traités par le groupe témoin. La plupart des élévations des transaminases ALAT sont survenues lors de l'utilisation concomitante de méthotrexate. Il n'y a pas eu d'élévations des transaminases ALAT ≥ 3 x LSN dans l'essai clinique de phase 3 d'Humira chez des patients atteints d'arthrite juvénile idiopathique polyarticulaire.2 et
Dans les essais cliniques contrôlés de phase 3 d'Humira chez des patients atteints de la maladie de Crohn et de rectocolite hémorragique avec des périodes de suivi allant de 4 à 52 semaines, des élévations d'ALAT supérieures ou égales à 3 fois la valeur normale maximale sont survenues chez 0,9 % des patients traités par Humira. patients et 0,9 % des patients traités par le groupe témoin.
Dans l'étude de phase 3 d'Humira chez des patients pédiatriques atteints de la maladie de Chron, qui a évalué l'innocuité et l'efficacité de deux schémas posologiques en fonction du poids pour le traitement d'entretien après un traitement d'induction en fonction du poids jusqu'à 52 semaines, des taux d'ALAT 3 x LSN ont été trouvés chez 2,6% de tous les patients exposés à un traitement concomitant avec des immunosuppresseurs de base.
Dans les études cliniques, dans toutes les indications, les patients présentant des taux élevés de transaminases étaient asymptomatiques et, dans la plupart des cas, les élévations étaient transitoires et ont disparu pendant le traitement. Cependant, des cas post-commercialisation d'insuffisance hépatique ainsi que des troubles hépatiques moins sévères pouvant précéder une insuffisance hépatique, tels que l'hépatite, y compris l'hépatite auto-immune, ont également été rapportés chez des patients traités par l'adalimumab.
Traitement concomitant par azathioprine / 6-mercaptopurine
Dans les études sur la maladie de Crohn chez l'adulte, des incidences plus élevées d'événements indésirables liés à des infections graves et à des tumeurs malignes ont été observées avec l'association d'Humira et d'azathioprine/6-mercaptopurine par rapport à Humira seul.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés survenant après l'autorisation du médicament est importante, car elle permet un suivi continu du rapport bénéfice/risque du médicament.Les professionnels de santé sont invités à déclarer tout effet indésirable suspecté via l'Agence italienne du médicament. , site web : http://www.agenziafarmaco.gov.it/it/responsabili.
04.9 Surdosage
Aucune toxicité liée à la dose n'a été observée au cours des études cliniques. La dose la plus élevée évaluée était des doses multiples de 10 mg/kg par voie intraveineuse; cette dose équivaut à environ 15 fois la dose recommandée.
05.0 PROPRIÉTÉS PHARMACOLOGIQUES
05.1 Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Agents immunosuppresseurs sélectifs. Code ATC : L04AB04
Mécanisme d'action
L'adalimumab se lie sélectivement au TNF et neutralise sa fonction biologique en bloquant son interaction avec les récepteurs membranaires du TNF, p55 et p75.
L'adalimumab module également les réponses biologiques induites ou régulées par le TNF, y compris les modifications des niveaux de molécules d'adhésion responsables de la migration des leucocytes (ELAM-1, VCAM-1 et ICAM-1 avec une CI50 de 0,1-0, 2 nM).
Effets pharmacodynamiques
Après traitement par Humira, une diminution rapide des protéines de la phase aiguë, des indices d'inflammation (protéine C réactive -PCR, vitesse de sédimentation érythrocytaire -VES) et des cytokines sériques (IL-6) a été observée chez les patients atteints de polyarthrite rhumatoïde par rapport à la base. Les taux sériques de métalloprotéinases matricielles (MMP-1 et MMP-3), impliquées dans le remodelage tissulaire responsable de la destruction du cartilage, ont également diminué après l'administration d'Humira. Les patients traités par Humira ont généralement montré une amélioration des signes biochimiques d'inflammation chronique.
Une diminution rapide des taux de protéine C réactive (CRP) a également été observée après un traitement par Humira chez des patients atteints d'arthrite juvénile idiopathique polyarticulaire, de maladie de Crohn et de rectocolite hémorragique. Chez les patients atteints de la maladie de Crohn, une diminution du nombre de cellules exprimant des marqueurs inflammatoires dans le côlon incluant une diminution significative de l'expression du TNFα a été observée.Des études endoscopiques de la muqueuse intestinale ont montré une cicatrisation de la muqueuse chez les patients traités par adalimumab.
Efficacité et sécurité cliniques
La polyarthrite rhumatoïde
Humira a été évalué chez plus de 3 000 patients dans tous les essais cliniques sur la polyarthrite rhumatoïde.L'efficacité et la sécurité d'Humira ont été évaluées dans cinq études randomisées, en double aveugle et bien contrôlées. Certains patients ont été traités jusqu'à 120 mois.
L'étude I sur la PR a été menée chez 271 patients ≥ 18 ans atteints de polyarthrite rhumatoïde active modérée à sévère réfractaire à au moins un DMARD incluant le méthotrexate à des doses allant de 12,5 à 25 mg (10 mg en cas d'intolérance au méthotrexate) par semaine et dont la dose de métatrexate est resté constant à 10-25 mg par semaine. Humira 20, 40 ou 80 mg ou un placebo a été administré toutes les deux semaines pendant 24 semaines.
L'étude AR II a étudié 544 patients âgés de ≥ 18 ans atteints de polyarthrite rhumatoïde active modérée à sévère avec une réponse insuffisante à au moins un DMARD. Des doses de 20 ou 40 mg d'Humira ont été administrées par injection sous-cutanée toutes les deux semaines avec un placebo toutes les deux semaines, ou toutes les semaines pendant 26 semaines ; le placebo a été administré chaque semaine pendant la même durée. L'utilisation d'autres ARMM n'était pas autorisée.
L'étude AR III a porté sur 619 patients, ≥ 18 ans, atteints de polyarthrite rhumatoïde active modérée à sévère avec une réponse inadéquate au traitement par méthotrexate à des doses allant de 12,5 à 25 mg, ou intolérants à 10 mg de méthotrexate chaque semaine. Dans cette étude, 3 groupes ont été constitués. Le premier a reçu des injections de placebo chaque semaine pendant 52 semaines. Le second a reçu Humira 20 mg par semaine pendant 52 semaines, tandis que le troisième a reçu Humira 40 mg toutes les deux semaines et des injections de placebo toutes les deux semaines. À l'issue des 52 premières semaines, 457 patients ont été inclus dans une phase d'extension en ouvert au cours de laquelle Humira/MTX a été administré à une dose de 40 mg toutes les deux semaines pendant une période allant jusqu'à 10 ans.
L'étude AR IV a d'abord évalué la sécurité d'Humira chez 636 patients atteints de polyarthrite rhumatoïde active modérée à sévère âgés de ≥ 18 ans. La population à l'étude était constituée à la fois de patients n'ayant jamais été traités par DMARD et de patients ayant poursuivi un traitement antirhumatismal préexistant à condition que celui-ci soit stable pendant au moins 28 jours. Ces traitements comprennent le méthotrexate, le léflunomide, l'hydroxychloroquine, la sulfasalazine et/ou les sels d'or. Les patients ont été randomisés pour recevoir Humira 40 mg ou un placebo toutes les deux semaines pendant 24 semaines.
L'étude AR V a évalué 799 patients adultes qui n'avaient jamais été traités par méthotrexate auparavant et avaient une polyarthrite rhumatoïde active précoce modérée à sévère (durée moyenne de la maladie inférieure à 9 mois). Cette étude a évalué l'efficacité d'Humira 40 mg administré toutes les deux semaines en association avec le méthotrexate, d'Humira 40 mg administré en monothérapie toutes les deux semaines et le méthotrexate seul pour réduire les signes et symptômes de la maladie et l'indice de progression des lésions articulaires causées par la polyarthrite rhumatoïde. pendant 104 semaines.
Le critère d'évaluation principal des études AR I, II, III et les critères d'évaluation secondaires de l'AR IV étaient d'évaluer la proportion de patients obtenant une réponse ACR 20 à la semaine 24 ou 26. L'objectif principal de l'étude AR V était d'évaluer la pourcentage de patients ayant obtenu une réponse ACR 50 à la semaine 52. De plus, les études AR III et V avaient pour objectif principal de démontrer l'inhibition de la progression de la maladie (par des examens radiographiques) à la semaine 52. L'étude AR III avait également pour objectif principal de démontrer une meilleure qualité de vie.
Réponse ACR
Les pourcentages de patients traités par Humira ayant obtenu des réponses ACR 20, 50 et 70 étaient comparables dans les études AR I, II et III. Les résultats du traitement par 40 mg toutes les deux semaines sont résumés dans le tableau 3.
Dans les études I-IV sur la PR, tous les paramètres évalués pour la définition de la réponse ACR (nombre d'articulations douloureuses et enflées, évaluation de l'activité de la maladie par le médecin et le patient, évaluation de la douleur par le patient, indice d'incapacité - HAQ) et les valeurs de CRP (mg/dL) s'est améliorée à 24 ou 26 semaines par rapport au placebo.Dans l'étude AR III, ces améliorations se sont maintenues sur 52 semaines.
Dans la phase d'extension en ouvert de l'étude AR III, la plupart des patients ayant présenté une réponse ACR ont maintenu la réponse lorsqu'ils ont poursuivi le traitement pendant 10 ans. Sur un total de 207 patients randomisés pour recevoir Humira 40 mg toutes les deux semaines, 114 ont continué à prendre Humira 40 mg toutes les deux semaines pendant 5 ans. Parmi ceux-ci, 86 patients (75,4 %) ont eu des réponses ACR 20 ; 72 patients (63,2 %) ont eu des réponses ACR 50 ; et 41 patients (36 %) ont présenté des réponses ACR 70. Sur un total de 207 patients, 81 ont poursuivi le traitement par Humira 40 mg toutes les deux semaines pendant 10 ans. Parmi ceux-ci, 64 patients (79,0 %) ont eu des réponses ACR 20 ; 56 patients (69,1 %) ont eu des réponses ACR 50 ; et 43 patients (53,1 %) avaient des réponses ACR 70.
Dans l'étude IV sur la PR, la réponse ACR 20 des patients traités par Humira en association avec un traitement conventionnel ≤ était statistiquement significativement meilleure que chez les patients traités par placebo associé à des médicaments traditionnels (p
Dans les études I-IV sur la PR, les patients traités par Humira ont obtenu des réponses ACR 20 et 50 statistiquement significativement plus élevées que le placebo dès 1 à 2 semaines après le début du traitement.
Dans l'étude V sur la PR, chez des patients atteints de polyarthrite rhumatoïde précoce qui n'avaient jamais été traités par méthotrexate, l'association Humira/méthotrexate a entraîné des réponses ACR plus rapides et significativement plus élevées que le méthotrexate en monothérapie et Humira en monothérapie à la semaine 52 et ces réponses ont persisté pendant 104 semaines ( voir tableau 4).
À la semaine 52, 42,9 % des patients recevant l'association Humira/méthotrexate ont obtenu une rémission clinique (DAS28
Réponse radiologique
Dans l'étude AR III, où les patients traités par Humira avaient une durée moyenne de la maladie d'environ 11 ans, les dommages structurels ont été évalués par radiographie et exprimés sous la forme d'une modification du score de Sharp total (TSS) modifié et des composants associés, de l'érosion et du rétrécissement de l'espace articulaire ( JSN).
Dans l'extension en ouvert de l'étude AR III, la réduction du taux de progression des dommages structurels est maintenue pendant 8 et 10 ans dans un sous-groupe de patients.A 8 ans, 81 des 207 patients initialement traités par Humira 40 mg tous les Les autres semaines ont été évaluées radiologiquement à 5 ans. Parmi ceux-ci, 48 patients n'ont présenté aucune progression des dommages structurels définis par un changement de mTSS de 0,5 ou moins par rapport à la valeur initiale. À 10 ans, 79 des 207 patients traités initialement par Humira 40 mg tous les deux semaine a été radiologiquement évaluée. Parmi ceux-ci, 40 patients n'ont pas montré de progression des dommages structurels définis par un changement de mTSS de 0,5 ou moins par rapport à la ligne de base.
au méthotrexate
b Intervalle de confiance à 95 % pour les différences dans les changements d'indice entre le méthotrexate et l'Humira.
c Sur la base d'une analyse de classement
dJoint Space Narrowing (réduction de l'espace entre les articulations)
Dans l'étude AR V, les dommages structuraux articulaires ont été évalués par radiographie et sont exprimés en termes de changement du score de Sharp total modifié (voir tableau 6).
Après 52 semaines et 104 semaines de traitement, la proportion de patients sans progression (variation par rapport à l'inclusion du score de Sharp total modifié & ≤ 0,5) était significativement plus élevée avec l'association Humira/méthotrexate (63,8 % et 61,2 %, respectivement) par rapport à méthotrexate en monothérapie (37,4 % et 33,5 %, respectivement, p
Qualité de vie et fonction physique
La qualité de vie et la fonction physique ont été évaluées à l'aide de l'indice d'incapacité obtenu par le biais du questionnaire d'évaluation de la santé (HAQ), dans quatre études originales, adéquates et bien contrôlées, et était l'un des critères d'évaluation principaux de l'étude AR III à la semaine 52. Tous Humira les régimes dans les quatre études ont montré des améliorations statistiquement significatives de l'indice d'incapacité du placebo HAQ entre la ligne de base et le mois 6 par rapport au placebo et dans l'étude AR III, le même résultat a été observé à la semaine 52. L'analyse de l'état de santé général, évaluée par le Short Form Health Survey (SF -36) dans les quatre études, étaye ces conclusions pour tous les schémas posologiques d'Humira avec des résultats statistiquement rapportés significatifs en ce qui concerne les indices d'activité physique, de douleur et de bien-être, enregistrés avec Humira 40 mg per se ttimes alternent. Une diminution statistiquement significative de la sensation de fatigue comme le montrent les indices de l'évaluation fonctionnelle relative au traitement des maladies chroniques (FACIT) a été retrouvée dans les trois études dans lesquelles elle a été évaluée (études AR I, III, IV).
Dans l'étude AR III, la majorité des sujets qui ont obtenu une amélioration de la fonction physique et qui ont poursuivi le traitement ont maintenu une amélioration pendant 520 semaines (120 mois) de traitement en ouvert. L'amélioration de la qualité de vie a été mesurée jusqu'à la semaine 156 (36 mois) et l'amélioration s'est maintenue dans le temps.
Dans l'étude AR V, l'indice d'incapacité évalué par le HAQ et la composante physique du SF 36 ont démontré une amélioration supérieure (p
Arthrite idiopathique juvénile
Arthrite juvénile idiopathique polyarticulaire (AJIp)
La sécurité et l'efficacité d'Humira ont été évaluées dans deux études (AJIp I et II) chez des enfants atteints d'arthrite juvénile idiopathique polyarticulaire ou polyarticulaire active, qui présentaient différents types d'AJI (le plus souvent une polyarthrite à facteur rhumatoïde négatif ou positif et une oligoarthrite étendue).
AJI I
La sécurité et l'efficacité d'Humira ont été évaluées dans une étude multicentrique, randomisée, en double aveugle, en groupes parallèles chez 171 enfants (âgés de 4 à 17 ans) atteints d'arthrite juvénile idiopathique polyarticulaire (AJI). = OL LI, les patients ont été stratifiés en deux groupes, le groupe MTX (méthotrexate) et le groupe MTX non traité. Le bras non traité par MTX n'avait jamais été traité par MTX auparavant ou avait arrêté de prendre MTX au moins deux semaines avant l'administration du médicament à l'étude. Les patients ont reçu des doses constantes d'anti-inflammatoires non stéroïdiens (AINS) et/ou de prednisone (≤0,2 mg/kg/jour ou un maximum de 10 mg/jour) Pendant la phase OL LI, tous les patients ont reçu Humira 24 mg/jour. m2 jusqu'à une dose maximale de 40 mg toutes les deux semaines pendant 16 semaines La répartition des patients selon l'âge et la dose minimale, moyenne et maximale administrée au cours de la phase OL LI sont présentées dans le tableau 7.
Tableau 7
Répartition des patients selon l'âge et la dose d'adalimumab administrée pendant la phase OL LI
Les patients qui ont démontré une réponse ACR30 pédiatrique à la semaine 16 étaient éligibles pour être éligibles à la randomisation dans la phase en double aveugle (DB) de l'étude et ont reçu Humira 24 mg/m2 jusqu'à un maximum de 40 mg ou un placebo toutes les deux semaines pendant 32 semaines ou jusqu'à la poussée de la maladie. Les critères de définition de l'exacerbation de la maladie ont été définis sur la base d'une aggravation supérieure ou égale à 30 % (≥ 30 %) par rapport à la valeur de base de 3 ou plus des 6 critères principaux du « Noyau pédiatrique ACR », en la présence de 2 articulations actives ou plus, et sur la base d'une amélioration supérieure à 30 % dans un maximum de 1 des 6 critères ci-dessus. Après 32 semaines ou lorsque la poussée de la maladie s'est produite, les patients ont été considérés comme ayant les exigences nécessaires pour être admis en phase d'extension en ouvert.
Tableau 8
Réponse PedACR30 au cours de l'étude JIA
a Les réponses Ped ACR 30/50/70 à la semaine 48 étaient significativement supérieures à celles obtenues chez les patients traités par placebo
bp = 0,015
cp = 0,031
Parmi ceux qui ont répondu au traitement à la semaine 16 (n = 144), les réponses Ped ACR 30/50/70/90 ont été maintenues jusqu'à six ans pendant la phase OLE chez les patients qui ont reçu Humira pendant toute la durée du studio. Au total, 19 sujets, dont 11 du groupe central âgés de 4 à 12 ans et 8 du groupe d'âge central 13 à 17 ans, ont été traités pendant 6 ans ou plus.
Les réponses globales étaient généralement meilleures et moins de patients ont développé des anticorps lorsqu'ils étaient traités avec l'association Humira et MTX par rapport à Humira administré seul. Compte tenu de ces résultats, l'utilisation d'Humira est recommandée en association avec le MTX et en monothérapie chez les patients pour lesquels l'utilisation de MTX n'est pas recommandée (voir rubrique 4.2).
pAJI II
La sécurité et l'efficacité d'Humira ont été évaluées dans une étude multicentrique en ouvert chez 32 enfants (2-2 surface corporelle d'Humira jusqu'à un maximum de 20 mg toutes les deux semaines en dose unique sous-cutanée pendant au moins 24 semaines. dans l'étude, la plupart des sujets utilisaient simultanément du MTX, certains sujets déclarant avoir utilisé des corticostéroïdes ou des anti-inflammatoires non stéroïdiens (AINS).
À la semaine 12 et à la semaine 24, la réponse PedACR30 était de 93,5 % et 90,0 %, respectivement, en utilisant l'approche des données observées. Les proportions de sujets avec PedACR50 / 70/90 à la semaine 12 et à la semaine 24 étaient respectivement de 90,3 % / 61,3 % / 38,7 % et 83,3 % / 73,3 % / 36,7 %. -étude d'extension de l'étiquette. Au total, ≤ 20 sujets ont été traités pendant 60 semaines ou plus.
Arthrite associée à une enthésite
La sécurité et l'efficacité d'Humira ont été évaluées dans une étude multicentrique, randomisée, en double aveugle chez 46 patients pédiatriques (âgés de 6 à 17 ans) atteints d'enthésite modérée associée à l'arthrite. Les patients ont été randomisés pour recevoir ou Humira 24 mg/m2 de surface corporelle. jusqu'à un maximum de 40 mg, ou un placebo toutes les deux semaines pendant 12 semaines.La période en double aveugle a été suivie d'une période d'étude en ouvert, au cours de laquelle les patients ont reçu Humira 24 mg/m2 de surface corporelle, jusqu'à un maximum de 40 mg par voie sous-cutanée toutes les deux semaines, pendant 192 semaines supplémentaires. Le critère d'évaluation principal était la variation en pourcentage du nombre d'articulations actives atteintes d'arthrite entre l'inclusion et la semaine 12. (gonflement non dû à une déformation ou des articulations avec perte de mouvement plus douleur et/ou douleur), qui a été obtenue avec une diminution moyenne en pourcentage de -62,6% (variation moyenne en pourcentage - 88,9%) chez les patients dans le groupe Humira par rapport à -11,6 % (variation moyenne en pourcentage - 50,0 %) chez les patients du groupe placebo. L'amélioration du nombre d'articulations actives atteintes d'arthrite s'est maintenue pendant la période en ouvert de l'étude jusqu'à la semaine 52. Bien que non statistiquement significative, la plupart des patients ont présenté une amélioration clinique du critère d'évaluation secondaire, tel que le nombre de sites d'enthésite, douleur nombre d'articulations (TJC), nombre d'articulations enflées (SJC), réponse ACR 50 pédiatrique et réponse ACR 70 pédiatrique.
Spondylarthrite axiale
Spondylarthrite ankylosante (SA)
L'administration d'Humira 40 mg pris toutes les deux semaines par 393 patients a été évaluée dans deux études randomisées en double aveugle contre placebo de 24 semaines chez des sujets atteints de spondylarthrite ankylosante active (où le score moyen de base de « activité de la maladie [Bath Ankylosing Spondylitis Disease Activity (BASDAI)] était égal à 6,3 dans tous les groupes analysés) qui ont développé une réponse inadéquate au traitement conventionnel. Soixante-dix-neuf patients (20,1 %) ont été traités par un traitement concomitant par DMARD et 37 patients (9,4 %) par des glucocorticoïdes. La période en aveugle a été suivie d'une période en ouvert au cours de laquelle les patients ont reçu 40 mg d'Humira toutes les deux semaines par voie sous-cutanée pendant une période supplémentaire pouvant aller jusqu'à 28 semaines. 12, ou la semaine 16 ou la semaine 20, ont été donnés ont utilisé 40 mg d'adalimumab par voie sous-cutanée comme traitement de secours précoce en ouvert toutes les deux semaines et ont ensuite été traités comme des non-répondeurs dans des analyses statistiques en double aveugle.
Dans une étude AS I plus vaste dans laquelle 315 patients ont été analysés, les résultats ont montré une amélioration statistiquement significative des signes et symptômes de la spondylarthrite ankylosante chez les patients traités par Humira par rapport aux patients traités par placebo.
La réponse significative a été observée pour la première fois à la semaine 2 et s'est maintenue sur une période de 24 semaines (tableau 9).
Tableau 9
Réponses d'efficacité dans l'étude sur la spondylarthrite ankylosante contrôlée par placebo - Étude I Réduction des signes et des symptômes
***, ** Statistiquement significatif sur p
a Évaluations de la spondylarthrite ankylosante
Indice d'activité de la maladie de la spondylarthrite ankylosante bBath
Les patients traités par Humira ont présenté une amélioration significativement plus élevée à la semaine 12, qui s'est maintenue pendant toute la durée du traitement jusqu'à la semaine 24, tant dans le SF36 que dans le questionnaire sur la qualité de vie de la spondylarthrite ankylosante (ASAQoL).
Des tendances similaires (pas toutes statistiquement significatives) ont été observées dans une étude AS II randomisée, en double aveugle et contrôlée par placebo chez 82 patients adultes atteints de spondylarthrite ankylosante active.
Spondylarthrite axiale sans signe radiographique de SA
Humira, administré à une dose de 40 mg toutes les deux semaines, a été évalué chez 185 patients atteints de spondylarthrite axiale non radiographique active dans une étude randomisée de 12 semaines, en double aveugle, contrôlée par placebo (la valeur de base moyenne de l'activité de la maladie [ Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)] était de 6,4 pour les patients traités par Humira et de 6,5 pour les patients traités par placebo) qui présentaient une réponse inadéquate ou une intolérance à un ou plusieurs AINS, ou une contre-indication aux AINS.
Trente-trois patients (18 %) ont été traités simultanément par des médicaments antirhumatismaux de fond et 146 patients (79 %) par des AINS au départ. La période en double aveugle a été suivie d'une période en ouvert au cours de laquelle les patients ont reçu 40 mg d'Humira toutes les deux semaines par voie sous-cutanée pendant jusqu'à 144 semaines. Les résultats de la semaine 12 ont démontré une amélioration statistiquement significative des signes et symptômes de la spondylarthrite axiale non radiographique active chez les patients traités par Humira par rapport au placebo (Tableau 10).
Tableau 10
Réponse d'efficacité dans une étude contrôlée contre placebo dans la spondylarthrite axiale - Réduction des signes et des symptômes
La qualité de vie liée à la santé et la fonction physique ont été évaluées à l'aide des questionnaires HAQ-S et SF-36. Humira a démontré une amélioration statistiquement significative plus importante par rapport à l'inclusion que le placebo du score total HAQ-S et du score de composante physique SF-36 à la semaine 12.
Arthrite psoriasique
Humira, administré à une dose de 40 mg toutes les deux semaines, a été étudié chez des patients atteints de rhumatisme psoriasique actif modéré à sévère dans deux études contrôlées versus placebo, PsA I et II. Au cours de l'étude PsA I de 24 semaines, 313 patients adultes ayant eu une réponse inadéquate au traitement anti-inflammatoire non stéroïdien ont été traités et parmi eux, environ 50 % prenaient du méthotrexate. Dans l'étude PsA II de 12 semaines, 100 patients ayant eu une réponse inadéquate au traitement par DMARD ont été traités. À la fin des deux études, 383 patients ont été inclus dans une étude d'extension en ouvert et ont reçu Humira 40 mg toutes les deux semaines.
En raison du nombre limité de patients étudiés, les preuves de l'efficacité d'Humira chez les patients atteints de rhumatisme psoriasique de type spondylarthrite ankylosante sont insuffisantes.
Tableau 11
Réponses ACR dans les études contrôlées par placebo dans les cas de rhumatisme psoriasique (pourcentage de patients)
Les réponses ACR dans le PsA I étaient similaires avec et sans traitement concomitant par méthotrexate.
Les réponses ACR dans l'étude d'extension en ouvert ont été maintenues jusqu'à 136 semaines.
Les modifications radiologiques ont été évaluées dans les études sur le rhumatisme psoriasique. Des radiographies des mains, des poignets et des pieds ont été prises au départ et à la semaine 24, pendant la phase en double aveugle lorsque les patients étaient traités par Humira ou un placebo, et à la semaine 48, lorsque tous les patients ont été traités par Humira en ouvert, avec un score de Sharp total modifié (mTSS) qui incluait les articulations interphalangiennes distales (c'est-à-dire différent du score de Sharp total utilisé pour la polyarthrite rhumatoïde).
Le traitement par Humira, par rapport au traitement par placebo, a réduit le taux de progression des lésions articulaires périphériques, tel que mesuré par les modifications du score de Sharp total modifié par rapport à l'inclusion (moyenne ± écart-type) 0,8 ± 2,5 dans le groupe placebo (à la semaine 24) contre 0,0 ± 1,9 (p
Chez les patients traités par Humira sans progression des lésions radiologiques de l'inclusion à la semaine 48 (n = 102), 84 % ont continué à ne présenter aucune progression des lésions radiologiques au cours des 144 semaines de traitement.
Les patients traités par Humira ont présenté une amélioration statistiquement significative de la fonction physique à la semaine 24 par rapport aux patients traités par placebo, tel qu'évalué par le HAQ et la Short Form Health Survey (SF 36).L'amélioration de la fonction physique s'est poursuivie jusqu'à la semaine 136 dans l'étude d'extension en ouvert .
Psoriasis
L'innocuité et l'efficacité d'Humira ont été étudiées chez des patients adultes atteints de psoriasis en plaques chronique (BSA ≥10 % et indice de surface et de gravité du psoriasis (PASI) ≥12 ou ≥10) candidats à un traitement systémique ou à une photothérapie. 73 % des patients admis dans les études de psoriasis I et II avaient déjà reçu un traitement systémique ou de photothérapie. La sécurité et l'efficacité d'Humira ont également été étudiées chez des patients adultes atteints de psoriasis chronique à modéré à sévère avec plaque concomitante localisée de la main et/ou du pied. psoriasis, qui étaient candidats à un traitement systémique dans une étude randomisée en double aveugle (étude de phase III sur le psoriasis).
L'étude I sur le psoriasis (REVEAL) a évalué 1 212 patients au cours de trois périodes de traitement. Au cours de la période A, les patients ont reçu soit un placebo, soit Humira à une dose initiale de 80 mg, suivie d'une dose de 40 mg, toutes les deux semaines, en commençant la semaine suivant la dose initiale. au moins une réponse PASI 75 (dont le score PASI s'est amélioré d'au moins 75 % par rapport à l'inclusion) a été admise à la période B et a reçu une dose d'Humira égale à 40 mg, toutes les deux semaines, en ouvert. semaine 33 et qui avaient été initialement randomisés pour recevoir un traitement actif au cours de la période A ont été de nouveau randomisés au cours de la période C pour recevoir 40 mg d'Humira, toutes les deux semaines, ou un placebo, pendant 19 semaines supplémentaires. Dans tous les groupes de traitement, le score PASI moyen au départ était de 18,9 et le score du médecin était « L'évaluation globale (PGA) était « modérée » chez 53 % des sujets inclus, « sévère » chez 41 % et « très sévère » chez 6,6 %.
L'étude II sur le psoriasis (CHAMPION) a comparé l'efficacité et l'innocuité d'Humira au méthotrexate et au placebo chez 271 patients. Sur une période de 16 semaines, les patients ont reçu un placebo ou du méthotrexate avec une dose initiale de 7, 5 mg par la suite augmentée jusqu'à la semaine 12 et avec un maximum de 25 mg, ou Humira à la dose initiale de 80 mg suivie de la dose de 40 mg administrée toutes les deux semaines (en commençant la semaine suivant la dose initiale). Il n'y a pas de données disponibles comparant Humira et le méthotrexate au-delà de 16 semaines de traitement. Chez les patients traités par méthotrexate qui ont obtenu une réponse ≥PASI 50 à la semaine 8 et/ou à la semaine 12, aucune augmentation de dose n'a été effectuée. Dans tous les groupes de traitement, le score PASI initial moyen était de 19,7 et le score PGA initial était « léger » (
Les patients qui ont participé à toutes les études de phase 2 et de phase 3 sur le psoriasis ont été considérés comme éligibles pour participer à une étude d'extension en ouvert, où Humira a été administré pendant une période supplémentaire d'au moins 108 semaines.
Dans les études sur le psoriasis I et II, le critère d'évaluation principal était la proportion de patients ayant obtenu une réponse PASI 75 au départ à la semaine 16 (voir les tableaux 12 et 13).
Dans l'étude I sur le psoriasis, 28 % des patients randomisés pour recevoir le placebo à la semaine 33 après avoir obtenu une réponse PASI 75 par rapport à 5 % des patients qui ont poursuivi le traitement par Humira, p
Dans l'étude I sur le psoriasis, un total de 233 patients, y compris ceux qui ont obtenu une réponse PASI 75 à la semaine 16 et à la semaine 33 et qui ont poursuivi le traitement par Humira jusqu'à la semaine 52, ont poursuivi le traitement par Humira dans l'étude d'extension en ouvert. Chez ces patients, les taux de réponse PASI 75 et PGA correspondant à une rémission totale de la maladie ou à une persistance minimale de la maladie étaient respectivement de 74,7% et 59,0%, après 108 semaines supplémentaires de traitement en ouvert (pour un total de 160 semaines de traitement continu). . Dans une analyse réalisée sur tous les patients qui avaient arrêté l'étude en raison d'événements indésirables ou d'un manque d'efficacité, ou qui avaient augmenté la posologie et qui pour ces raisons étaient considérés comme ne répondant pas au traitement, de même les taux de réponse PASI 75 et PGA correspondant à la rémission totale de la maladie ou à la persistance minimale de la maladie étaient de 69,6 % et 55,7 %, respectivement, après 108 semaines supplémentaires de traitement en ouvert (pour un total de 160 semaines de traitement continu).
Au total, 347 patients, caractérisés par une réponse stable au traitement, ont participé à une étude d'extension en ouvert pour évaluer les effets de l'arrêt et de la reprise du traitement. Pendant la période de sevrage, les symptômes du psoriasis sont réapparus progressivement avec un délai médian de rechute (évoluant vers un stade PGA « modéré » ou pire) d'environ 5 mois. Aucun de ces patients n'a connu de phénomène de rebond pendant la période d'arrêt du traitement. Dans l'ensemble, 76,5 % des patients (218/285) qui sont entrés dans la phase de retraitement ont obtenu une réponse PGA correspondant à une rémission totale de la maladie ou à une maladie minimale après seize semaines de traitement, qu'ils aient ou non eu ou non des poussées de la maladie. pendant la période d'attente du médicament (69,1 % [123/178] et 88,8 % [95/107], respectivement, des patients qui ont eu ou non une poussée de la maladie pendant la période d'attente) .
Un profil de sécurité très similaire a été observé lors de la reprise du traitement à celui observé au cours de la période précédant l'arrêt du traitement.
Des améliorations significatives de l'indice de qualité de vie dermatologique (DLQI) ont été démontrées à la semaine 16 par rapport au départ par rapport au placebo (études I et II) et au méthotrexate (étude II). Dans l'étude I, les améliorations des scores des composantes physiques et mentales globales du SF-36 étaient également significatives par rapport au placebo.
Dans une étude d'extension en ouvert, parmi les patients qui, en raison d'une réponse PASI de moins de 50 %, ont reçu des augmentations de dose de 40 mg toutes les deux semaines à 40 mg toutes les semaines, évaluées à la semaine 12 après l'augmentation de la dose, 93 sur 349 (26,6 % ) a obtenu une réponse PASI 75.
L'étude de phase III sur le psoriasis (REACH) a comparé l'efficacité et l'innocuité d'Humira versus placebo chez 71 patients atteints de psoriasis en plaques chronique modéré à sévère et de psoriasis localisé des mains et/ou des pieds. toutes les deux semaines (en commençant la semaine suivant la dose initiale) ou un placebo pendant 16 semaines. À la semaine 16, une proportion statistiquement plus élevée de patients ayant reçu Humira a obtenu une réponse PGA correspondant à une rémission totale ou quasi totale de la maladie de la main et/ou du pied par rapport aux patients ayant reçu le placebo (30,6 %, respectivement). = 0,014]).
la maladie de Crohn
L'innocuité et l'efficacité d'Humira ont été évaluées chez plus de 1 500 patients atteints de la maladie de Crohn modérément à sévèrement active (indice d'activité de la maladie de Crohn (CDAI)) ≥ 220 et ≤ 450) dans le cadre d'essais randomisés, en double aveugle, contrôlés par placebo. L'administration concomitante de doses constantes d'aminosalicylates, de corticoïdes et/ou d'agents immunomodulateurs était autorisée et 80 % des patients ont continué à prendre au moins un de ces médicaments.
L'induction de la rémission clinique (définie comme CDAI
Le maintien de la rémission clinique a été évalué dans l'étude CD III (CHARM).Dans l'étude III sur la MC, 854 patients ont reçu en ouvert 80 mg d'Humira à la semaine 0 et 40 mg à la semaine 2. À la semaine 4, les patients ont été randomisés pour recevoir 40 mg toutes les deux semaines, 40 mg toutes les semaines ou un placebo. ; la durée totale de l'étude était de 56 semaines. Les patients qui ont présenté une réponse clinique adéquate (diminution du CDAI 70) à la semaine 4 ont été stratifiés et analysés séparément de ceux qui n'ont pas présenté de réponse clinique adéquate à la semaine 4. Une réduction progressive de la dose de corticoïdes après la semaine 8.
Les taux d'induction de rémission clinique et de réponse de l'étude CD I et de l'étude CD II sont présentés dans le tableau 14.
Des taux de rémission similaires ont été observés dans le groupe recevant une dose d'induction de 160/80 mg et 80/40 mg à la semaine 8 et des événements indésirables sont survenus plus fréquemment dans le groupe recevant une dose de 160/80 mg.
Dans l'étude III sur la MC, à la semaine 4, 58 % (499/854) des patients ont présenté une réponse clinique adéquate et ont été évalués dans l'analyse principale. Parmi les patients ayant présenté une réponse clinique adéquate à la semaine 4, 48 % avaient déjà été exposés à un traitement avec d'autres anti-TNF.Les pourcentages de maintien de la rémission et de la réponse clinique sont indiqués dans le tableau 15. Les résultats pour la rémission clinique sont restés relativement constants quelle que soit l'exposition antérieure aux médicaments anti-TNF.
À la semaine 56, les hospitalisations et les interventions chirurgicales liées à la maladie étaient statistiquement significativement réduites avec l'adalimumab par rapport au placebo.
Parmi les patients qui n'ont pas montré de réponse adéquate à la semaine 4, 43 % des patients traités par le traitement d'entretien par Humira ont présenté une réponse adéquate à la semaine 12, contre 30 % des patients traités par placebo. Ces résultats suggèrent que certains patients qui n'ont pas présenté de réponse adéquate à la semaine 4 bénéficient d'un traitement d'entretien continu jusqu'à la semaine 12. Le traitement poursuivi au-delà de 12 semaines n'a pas conduit à un nombre significativement plus élevé de réponses (voir rubrique 4.2).
117/276 patients de l'étude MC I et 272/777 patients des études MC II et III ont été suivis pendant au moins 3 ans sous traitement ouvert à l'adalimumab. 88 et 189 patients, respectivement, ont continué à maintenir une rémission clinique. La réponse clinique (CR-100) s'est maintenue chez 102 et 233 patients, respectivement.
Qualité de vie
Dans les études CD I et CD II, une amélioration statistiquement significative du score total 4 du questionnaire sur les maladies inflammatoires de l'intestin spécifiques à la maladie (IBDQ) a été obtenue à la semaine 4 chez les patients randomisés pour recevoir Humira 80/40 mg et 160/80 mg par rapport au placebo et a été observée. aux semaines 26 et 56 dans l'étude III sur la MC ainsi qu'entre les groupes de traitement Humira par rapport au groupe placebo.
La maladie de Crohn chez les patients pédiatriques
Humira a été testé dans une étude clinique multicentrique, randomisée et en double aveugle conçue pour évaluer l'efficacité et l'innocuité d'un traitement d'induction et d'entretien dose-dépendant en fonction du poids (maladie de Crohn (MC) modérée à sévère). définie par un indice d'activité de la maladie de Crohn pédiatrique (PCDAI) score > 30. Les sujets doivent avoir échoué au traitement conventionnel (y compris un corticostéroïde et/ou un immunomodulateur) de la MC. De plus, les sujets peuvent avoir perdu la réponse ou être intolérants à l'infliximab.
Tous les sujets ont reçu un traitement d'induction en ouvert avec une dose basée sur leur poids corporel à l'inclusion : 160 mg à la semaine 0 et 80 mg à la semaine 2 pour les sujets pesant ≥ 40 kg, et 80 mg et 40 mg, respectivement.
À la semaine 4, en fonction de leur poids corporel, les sujets ont été randomisés 1 : 1 pour recevoir les régimes d'entretien à faible dose ou à dose standard, comme indiqué dans le tableau 16.
Résultats d'efficacité
Le critère d'évaluation principal de l'étude était la rémission clinique à la semaine 26, définie par un score PCDAI & ≤ 10.
Les taux de rémission clinique et de réponse clinique (définis comme une réduction du score PCDAI d'au moins 15 points par rapport à la ligne de base) sont présentés dans le tableau 17. Les taux d'arrêt des corticostéroïdes ou des immunomodulateurs sont présentés dans le tableau 18.
Des augmentations statistiquement significatives (améliorations) de l'indice de masse corporelle et de la vitesse de croissance de la ligne de base aux semaines 26 et 52 ont été observées pour les deux groupes de traitement.
Des améliorations statistiquement et cliniquement significatives par rapport aux valeurs initiales des paramètres de qualité de vie (y compris IMPACT III) ont également été observées dans les deux groupes de traitement.
Rectocolite hémorragique
La sécurité et l'efficacité de doses multiples d'Humira ont été évaluées chez des patients adultes atteints de rectocolite hémorragique active modérée à sévère (score Mayo de 6 à 12 avec un sous-score endoscopique de 2 à 3) dans des études randomisées, en double aveugle, contrôlées par placebo.
Dans l'étude UC-I, 390 patients non préalablement traités par anti-TNF ont été randomisés pour recevoir un placebo aux semaines 0 et 2, 160 mg d'Humira à la semaine 0 suivis de 80 mg à la semaine 2 ou 80 mg à la semaine 0 de suivi. à la semaine 2. Après la semaine 2, les patients inclus dans les deux bras adalimumab ont reçu 40 mg toutes les deux semaines. La rémission clinique (définie par un score Mayo ≤ 2 sans sous-score > 1) a été évaluée à la semaine 8.
Dans l'étude UC-II, 248 patients ont pris Humira 160 mg à la semaine 0, 80 mg à la semaine 2 et 40 mg toutes les deux semaines, et 246 patients ont pris un placebo. L'induction de la rémission a été évaluée à la semaine 8 et le maintien de la rémission à la semaine 52.
Dans l'étude UC-I (18 % vs 9 %, respectivement, p = 0,031) et dans l'étude UC-II (17 % vs 9 %, respectivement, p = 0,019), les patients induits par Humira 160/80 mg ont obtenu une rémission clinique par rapport à placebo à la semaine 8 dans des pourcentages statistiquement significativement plus élevés. Dans l'étude UC-II, parmi les patients traités par Humira en rémission à la semaine 8, 21/41 (51 %) étaient en rémission à la semaine 52.
Les résultats globaux de l'étude de population UC-II sont présentés dans le tableau 19.
Tableau 19
Réponse muqueuse, rémission et cicatrisation dans l'étude UC-II (pourcentage de patients)
Parmi les patients qui ont répondu à la semaine 8, 47 % étaient en réponse clinique, 29 % étaient en rémission, 41 % avaient une cicatrisation de la muqueuse et 20 % étaient en rémission clinique sans stéroïdes pendant ≥ 90 jours à la semaine 52.
Environ 40 % des patients inclus dans l'UC-II avaient échoué à un traitement anti-TNF antérieur par infliximab. L'efficacité de l'adalimumab chez ces patients a été réduite par rapport à celle observée chez les patients non préalablement traités par anti-TNF. Parmi les patients en échec à un traitement anti-TNF antérieur, 3 % du bras placebo et 10 % du bras adalimumab ont obtenu une rémission à semaine 52.
Les patients inclus dans UC-I et UC-II ont eu la possibilité de participer à une étude ouverte à long terme étendue (UC-III). Après trois ans de traitement à base d'adalimumab, 75 % (301/402) étaient toujours en rémission clinique selon le score partiel de Mayo.
Taux d'hospitalisation
Au cours des 52 semaines des études UC-I et UC-II, une diminution des hospitalisations toutes causes confondues et des hospitalisations liées à la rectocolite hémorragique a été observée dans le groupe adalimumab par rapport au groupe placebo. Le nombre d'hospitalisations toutes causes confondues était de 0,18 sujet par an dans le groupe de traitement adalimumab par rapport au groupe placebo (0,26 sujet par an) et les données correspondantes d'hospitalisations liées à la rectocolite hémorragique étaient de 0,12 sujet par an contre 0,22 sujet par an.
Qualité de vie
Dans l'étude UC-II, le traitement par l'adalimumab a entraîné une amélioration du score au questionnaire sur les maladies inflammatoires de l'intestin (IBDQ) spécifiques à la maladie.
Immunogénicité
La formation d'anticorps anti-adalimumab est associée à une augmentation de la clairance et à une diminution de l'efficacité de l'adalimumab. Il n'y a pas de corrélation évidente entre la présence d'anticorps anti-adalimumab et la survenue d'événements indésirables.
Les patients des études sur la polyarthrite rhumatoïde ont été dépistés à divers intervalles de temps pour les anticorps dirigés contre l'adalimumab au cours de la période de 6 à 12. Dans les études cliniques pivots, des anticorps dirigés contre l'adalimumab ont été détectés chez 5,5 % (58/1053) des patients traités par l'adalimumab, comparativement à avec 0,5 % (2/370) des patients traités par placebo. Chez les patients n'ayant pas reçu de méthotrexate de manière concomitante, l'incidence était de 12,4 %, contre 0,6 % lorsque l'adalimumab était utilisé en association avec le méthotrexate.
Chez les patients atteints d'arthrite juvénile idiopathique polyarticulaire âgés de 4 à 17 ans, des anticorps anti-adalimumab ont été identifiés chez 15,8 % des patients (27/171) traités par adalimumab. Chez les patients n'ayant pas reçu de méthotrexate avec Humira, l'incidence était de 25,6% (22/86) versus 5,9% (5/85) lorsque l'adalimumab était utilisé en association avec le méthotrexate.
Chez les patients atteints d'arthrite associée à l'enthésite, des anticorps anti-adalimumab ont été identifiés chez 10,9 % (5/46) des patients traités par l'adalimumab. Chez les patients n'ayant pas reçu de méthotrexate en concomitance avec Humira, l'incidence était de 13,6 % (3/22), contre 8,3 % (2/24) lorsque l'adalimumab était utilisé en association avec le méthotrexate.
Chez les patients atteints de rhumatisme psoriasique, des anticorps anti-adalimumab ont été identifiés chez 38/376 sujets (10 %) traités par adalimumab. Chez les patients ne recevant pas de traitement concomitant par méthotrexate, l'incidence était de 13,5 % (24/178 sujets) contre 7 % (14 des 198 sujets) lorsque l'adalimumab était utilisé en association avec le méthotrexate.
Chez les patients atteints de spondylarthrite ankylosante, des anticorps anti-adalimumab ont été identifiés chez 17/204 sujets (8,3 %) traités par adalimumab. Chez les patients ne recevant pas de traitement concomitant par méthotrexate, l'incidence était de 16/185 (8,6 %) contre 1/19 (5,3 %) lorsque l'adalimumab était utilisé en association avec le méthotrexate.
Chez les patients atteints de la maladie de Crohn, des anticorps anti-adalimumab ont été identifiés chez 7/269 sujets (2,6 %) et chez 19/487 sujets chez les patients atteints de rectocolite hémorragique.
Chez les patients atteints de psoriasis, des anticorps anti-adalimumab ont été identifiés chez 77/920 (8,4 %) sujets traités par adalimuman seul.
Chez les patients atteints de psoriasis en plaques traités au long cours en monothérapie par adalimumab qui ont participé à une étude de sevrage et de retraitement, le taux d'anticorps anti-adalimumab positifs après reprise du traitement (11 cas sur 482 sujets, 2,3 %) était similaire à celui observés avant l'arrêt du médicament (11 cas sur 590 sujets, 1,9 %).
Étant donné que les tests d'immunogénicité sont spécifiques au produit, la comparaison des quantités d'anticorps avec d'autres produits n'est pas appropriée.
Population pédiatrique
L'Agence européenne des médicaments a dérogé à l'obligation de soumettre les résultats des études menées avec Humira dans tous les sous-groupes de la population pédiatrique dans la polyarthrite rhumatoïde, le rhumatisme psoriasique et la spondylarthrite ankylosante, voir rubrique 4.2 pour les informations sur l'usage pédiatrique.
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats des études menées avec Humira dans un ou plusieurs sous-ensembles de la population pédiatrique dans la rectocolite hémorragique, voir rubrique 4.2 pour des informations sur l'usage pédiatrique.
05.2 Propriétés pharmacocinétiques
Absorption et distribution
Après l'administration sous-cutanée d'une dose unique de 40 mg, l'absorption et la distribution de l'adalimumab étaient lentes, les concentrations sériques maximales survenant environ 5 jours après l'administration.La biodisponibilité absolue moyenne de l'adalimumab dans les trois études après une dose sous-cutanée de 40 mg était de 64. Après des doses intraveineuses uniques de 0,25 à 10 mg/kg, les concentrations étaient proportionnelles à la dose.Après des doses de 0,5 mg/kg (≈40 mg), la clairance variait de 11 à 15 mL/heure, le volume de distribution (Vss) variait de 5 à 6 litres, et la demi-vie moyenne de la phase finale était d'environ deux semaines. Les concentrations d'adalimumab dans le liquide synovial chez divers patients atteints de polyarthrite rhumatoïde variaient de 31 à 96 % de celles dans le sérum.
Après l'administration sous-cutanée de 40 mg d'adalimumab toutes les deux semaines chez des patients adultes atteints de polyarthrite rhumatoïde (PR), les concentrations minimales étaient en moyenne d'environ 5 mcg/mL (sans traitement concomitant par méthotrexate) et de 8 à 9 mcg/mL (en association avec le méthotrexate). les taux sériques d'adalimumab à l'équilibre après des doses sous-cutanées de 20, 40 et 80 mg toutes les 2 semaines ou toutes les semaines ont augmenté de manière presque dose-dépendante.
Suite à l'administration d'adalimumab à 24 mg/m2 (jusqu'à un maximum de 40 mg) par voie sous-cutanée toutes les deux semaines à des patients âgés de 4 à 17 ans atteints d'arthrite juvénile idiopathique polyarticulaire (AJI), la concentration sérique minimale moyenne initiale d'adalimumab (valeurs mesurées à partir de entre la semaine 20 et la semaine 48) était de 5,6 ± 5,6 mcg/mL (102 % CV) avec l'adalimumab sans utilisation concomitante de méthotrexate et de 10,9 ± 5,2 mcg/mL (47,7 % CV) avec méthotrexate administré de façon concomitante.
Chez les patients atteints d'AJI polyarticulaire âgés de 2 à 2 ans, la concentration sérique minimale à l'équilibre de l'adalimumab était de 6,0 ± 6,1 g/mL (101 % CV) avec l'adalimumab sans utilisation concomitante de méthotrexate et de 7,9 ± 5,6 mcg/mL (71,2 % CV) lorsqu'il est co-administré avec le méthotrexate.
Après administration sous-cutanée de 24 mg/m2 (jusqu'à un maximum de 40 mg) toutes les deux semaines à des patients âgés de 6 à 17 ans atteints d'arthrite associée à l'enthésite, les concentrations sériques minimales moyennes d'adalimumab au stade d'état d'équilibre (valeurs mesurées à la semaine 24) étaient de 8,8 ± 6,6 mcg/mL avec l'adalimumab sans utilisation concomitante de méthotrexate et de 11,8 ± 4,3 mcg/mL en cas de co-administration avec le méthotrexate.
Chez les patients atteints de psoriasis, la concentration minimale s'est avérée en moyenne d'environ 5 mcg/mL pendant le traitement avec une dose de 40 mg d'adalimumab administrée toutes les deux semaines en monothérapie.
Chez les patients atteints de la maladie de Crohn, la dose de charge d'Humira 80 mg à la semaine 0 suivie d'Humira 40 mg à la semaine 2 a permis d'atteindre des concentrations sériques minimales d'adalimumab d'environ 5,5 mcg/mL pendant la période d'induction. Une dose de charge d'Humira 160 mg à la semaine 0 suivie d'Humira 80 mg à la semaine 2 a permis d'atteindre des concentrations sériques minimales d'adalimumab d'environ 12 mcg/mL pendant la période d'induction. Des taux d'équilibre moyens d'environ 7 mcg/mL ont été observés chez des patients atteints de la maladie de Crohn qui ont reçu une dose d'entretien de 40 mg d'Humira toutes les deux semaines.
Chez les patients pédiatriques atteints de MC modérée à sévère, la dose d'induction d'adalimumab en ouvert était de 160/80 mg ou 80/40 mg aux semaines 0 et 2, respectivement, en fonction du seuil de poids corporel à 40 kg. À la semaine 4, les patients ont été randomisés 1 : 1 en fonction du poids corporel dans le groupe de traitement à dose standard (40/20 mg toutes les deux semaines) ou à faible dose (20/10 mg toutes les deux semaines). Les concentrations sériques moyennes (± ET) des concentrations minimales d'adalimumab atteintes à la semaine 4 étaient de 15,7 ± 6,6 mcg/mL pour les patients ≥ 40 kg (160/80 mg) et de 10,6 ± 6,1 mcg/mL pour les sujets
Pour les patients restant sous traitement randomisé, les concentrations minimales moyennes (± ET) d'adalimumab à la semaine 52 étaient de 9,5 ± 5,6 mcg/mL pour le groupe à dose standard et de 3,5 ± 2,2 mcg/mL pour le groupe à faible dose. Les concentrations minimales moyennes ont été maintenues chez les patients qui ont continué à recevoir un traitement par adalimumab toutes les deux semaines pendant 52 semaines. Pour les patients qui ont augmenté la dose d'un régime toutes les deux semaines à une semaine ≤ les concentrations sériques moyennes (± ET) d'adalimumab à la semaine 52 étaient de 15,3 ± 11,4 g/mL (40/20 mg, par semaine) et de 6,7 ± 3,5 mcg/ ml (20/10 mg, par semaine).
Chez les patients atteints de rectocolite hémorragique, une dose de charge de 160 mg d'Humira à la semaine 0 suivie de 80 mg d'Humira à la semaine 2 a permis d'atteindre une concentration minimale d'adalimumab d'environ 12 µg/mL pendant la période d'induction. Des taux d'équilibre moyens d'environ 8 mcg/mL ont été observés chez des patients atteints de rectocolite hémorragique qui ont reçu une dose d'entretien d'Humira 40 mg toutes les deux semaines.
Élimination
Les analyses pharmacocinétiques de population sur un échantillon de plus de 1 300 patients atteints de PR ont montré une tendance à une augmentation apparente de la clairance de l'adalimumab à mesure que le poids corporel augmente. Après correction pour le poids corporel, les différences de sexe et d'âge ont un effet minime sur la clairance de l'adalimumab. L'adalimumab libre (non lié aux anticorps anti-adalimumab - AAA) était plus faible chez les patients présentant des titres mesurables d'AAA Humira n'a pas été étudié chez les patients présentant une insuffisance rénale ou hépatique.
Insuffisance hépatique ou rénale
Humira n'a pas été étudié chez les patients présentant une insuffisance rénale ou hépatique.
05.3 Données de sécurité précliniques
Les données non cliniques ne révèlent aucun risque particulier pour l'homme sur la base d'études de toxicité à dose unique ≤ de toxicité à doses multiples et d'études de génotoxicité.
Une étude de toxicité sur le développement embryo-fœtal / développement périnatal a été menée chez des singes cynomologiques aux doses de 0, 30 et 100 mg/kg (9-17 singes/groupe) ; cette étude n'a révélé aucun dommage fœtal causé par l'adalimumab. Les tests de cancérogénicité et les évaluations standard de la fertilité et de la toxicité postnatale n'ont pas été effectués en raison du manque de modèles appropriés pour un anticorps présentant une réactivité croisée limitée avec le TNF chez les rongeurs et le développement d'anticorps neutralisants chez les rongeurs.
06.0 INFORMATIONS PHARMACEUTIQUES
06.1 Excipients
Mannitol
Acide citrique monohydraté
Citrate de sodium
Phosphate monosodique dihydraté
Phosphate disodique dihydraté
Chlorure de sodium
Polysorbate 80
Hydroxyde de sodium
Eau pour préparations injectables.
06.2 Incompatibilité
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
06.3 Durée de validité
24mois
06.4 Précautions particulières de conservation
A conserver au réfrigérateur (2°C - 8°C). Ne pas congeler. Conservez le stylo prérempli dans l'emballage pour protéger le médicament de la lumière.
Un seul stylo Humira peut être conservé à des températures allant jusqu'à 25 °C maximum pendant une période ne dépassant pas 14 jours. Le stylo doit être protégé de la lumière et jeté s'il n'est pas utilisé pendant la période de 14 jours.
06.5 Nature du conditionnement primaire et contenu de l'emballage
Humira 40 mg solution injectable est contenu dans des stylos jetables préremplis prêts à l'emploi, contenant une seringue préremplie.La seringue à l'intérieur du stylo est en verre de type I avec un piston (caoutchouc bromobutyle) et une aiguille avec un capuchon (thermoplastique élastomère).
Emballage:
• 1 stylo prérempli avec 1 tampon d'alcool sous blister.
• 2 stylos pré-remplis, chacun avec 1 tampon d'alcool, sous blister.
• 4 stylos pré-remplis, chacun avec 1 tampon d'alcool, sous blister.
• 6 stylos pré-remplis, chacun avec 1 tampon d'alcool, sous blister.
Toutes les présentations peuvent ne pas être commercialisées.
06.6 Instructions d'utilisation et de manipulation
Humira 40 mg solution injectable ne contient aucun conservateur. Les médicaments non utilisés et les déchets de ce médicament doivent être éliminés conformément à la réglementation locale.
07.0 TITULAIRE DE L'AUTORISATION DE MISE SUR LE MARCHE
AbbVie Ltd
Virginité
SL6 4XE
Royaume-Uni
08.0 NUMÉRO D'AUTORISATION DE MISE SUR LE MARCHÉ
EU / 1/03/256/0071 stylo prérempli 0,8 ml + 1 tampon
035946072 / E
EU / 1/03/256/0082 stylos préremplis 0,8 ml + 2 tampons
035946084 / E
EU / 1/03/256/0094 stylos préremplis 0,8 ml + 4 tampons
035946096 / E
UE / 1/03/256/0106 stylos préremplis 0,8 ml + 6 écouvillons
035946108 / F
09.0 DATE DE PREMIÈRE AUTORISATION OU DE RENOUVELLEMENT DE L'AUTORISATION
Date de première autorisation : 08 septembre 2003
Date du dernier renouvellement : 08 septembre 2008
10.0 DATE DE RÉVISION DU TEXTE
09/2014