Maladie héréditaire, la SMA est due à des mutations du gène SMN1 ou du gène SMN2 dont le but est de produire une protéine qui sert à assurer la survie des motoneurones.
Il existe cinq formes différentes d'amyotrophie spinale : type 0, type 1, type 2, type 3 et type 4. Les trois premiers types sont très graves et entraînent la mort prématurée du patient ; le type 3 et le type 4 sont des variantes plus douces, qui affectent le niveau de vie du patient, mais sans provoquer de décès prématuré.
Un test génétique sur un échantillon de sang est nécessaire pour diagnostiquer l'AMS.
Actuellement, la thérapie de l'AMS repose principalement sur des traitements symptomatiques, visant à soulager les troubles et à contrôler les complications. Un remède existe, basé sur les principes de la thérapie génique, mais c'est une solution très coûteuse et applicable uniquement à certains patients.
, qui se manifeste par une atrophie et un affaiblissement consécutif des muscles squelettiques, et des difficultés motrices.
L'AMS est une affection qui peut entraîner la mort du patient à un jeune ou très jeune âge : les formes les plus graves de la maladie, en effet, affectent l'efficacité des muscles respiratoires et sont responsables d'épisodes d'insuffisance respiratoire ou de pneumonie avec un issue fatale.
Neurones moteurs et SMA

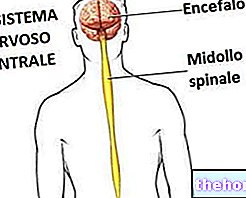
Les motoneurones, ou motoneurones, sont des cellules nerveuses qui naissent dans le système nerveux central (cerveau et moelle épinière) et qui, par l'intermédiaire de leurs prolongements (axones), contrôlent l'activité des muscles et des glandes.
Il existe deux types de motoneurones : les motoneurones supérieurs (ou premiers motoneurones) et les motoneurones inférieurs (ou seconds motoneurones).
Les motoneurones supérieurs proviennent du cerveau et dirigent l'activité des motoneurones inférieurs, qui surviennent principalement dans la moelle épinière et sont chargés de diriger l'activité des muscles squelettiques (ou somatiques), des muscles lisses (ou viscéraux), du muscle cardiaque et du cœur.
Les motoneurones des individus atteints d'AMS dégénèrent progressivement, provoquant « une atrophie musculaire due à l'inactivité qui, dans les cas les plus graves, entraîne une paralysie, une insuffisance respiratoire et la mort.
Épidémiologie : quelle est la fréquence de l'amyotrophie spinale ?
La SMA a une « incidence annuelle de 1 cas pour 10 000 nouvelles naissances.
5 et dont dépend la production de la protéine dite de survie des motoneurones (SMN).Comme le suggère le nom de la protéine produite par SMN1 et SMN2, la mutation de ces gènes prive les motoneurones d'une substance biologique essentielle à leur survie ; plus précisément, il réduit les niveaux de protéines : par exemple, en présence de mutations dans SMN1, les niveaux de protéines SMN chutent à 10-20% de la normale.
De toute évidence, l'absence de quantités adéquates de la protéine SMN détermine la dégénérescence progressive des motoneurones.
La perte des motoneurones interrompt la signalisation nerveuse qui permet de contrôler l'activité des muscles du corps humain ; ces derniers, du fait qu'ils ne sont plus utilisables, subissent un processus progressif d'atrophie et d'affaiblissement.
Saviez-vous que...
Le gène SMN2 est, pour la SMA, un gène modificateur de la maladie ; en fait, chez les patients porteurs d'une mutation dans SMN1 et qui ont, pour une raison quelconque, trois ou quatre copies du gène SMN2, la SMA survient sous une forme plus douce.
Amyotrophie spinale : types de mutation
Lorsque la SMA est due à une « altération de SMN1, dans 95 à 98 % des cas la mutation responsable consiste en une délétion du gène entier, alors que seulement dans 2 à 5 % dans une « anomalie de la séquence normale du gène.
Amyotrophie spinale : une maladie héréditaire
Dans la quasi-totalité des cas (98 %), l'anomalie génétique responsable de la SMA est héréditaire, c'est-à-dire que ce sont les parents du malade qui la transmettent.
2% des cas non héréditaires de SMA sont dus à une mutation de novo s'est produite à un stade très précoce du développement embryonnaire.
SMA et modèle d'héritage
Le modèle héréditaire de l'amyotrophie spinale est autosomique récessif, ce qui signifie que, pour hériter de la SMA, il est essentiel que les deux parents soient porteurs sains du défaut génétique de SMN1 ou SMN2 et que les deux parents le transmettent.
Dans le cas des maladies héréditaires autosomiques récessives telles que la SMA, la probabilité que les deux porteurs sains transmettent le défaut génétique à l'enfant, le rendant ainsi malade, est de 25 %, soit un cas sur 4.
Types de SMA
En fonction de l'âge d'apparition et de la gravité de la maladie, les experts reconnaissent cinq formes différentes d'amyotrophie spinale :
- SMA type 0 : c'est la forme la plus sévère de toutes. Elle se manifeste avant même la naissance avec une mobilité réduite du fœtus.
Les nourrissons survivent généralement quelques semaines après la naissance, même lorsqu'ils reçoivent une assistance respiratoire. - SMA type 1 : des formes qui surviennent au cours de la vie, c'est la plus sévère et la plus fréquente (environ 50 % des cas) ; il apparaît à un âge précoce, généralement au cours du sixième mois de vie.
En règle générale, c'est la cause de la mort déjà dans les premières années de la vie; rarement, pendant l'adolescence.
La mort survient généralement à la suite d'une « insuffisance respiratoire ou » d'une infection pulmonaire. - SMA type 2 : c'est la forme qui, par gravité, arrive en deuxième position ; généralement, il débute entre 7 et 18 mois de vie.
L'espérance de vie des personnes atteintes est plus importante que dans le cas précédent : les patients parviennent en effet à atteindre l'âge adulte. - SMA de type 3 : moins sévère que les deux précédentes, cette forme d'AMS survient généralement après 18 mois de vie (dans certains cas, elle peut également apparaître pendant l'enfance ou l'adolescence).
Elle implique des handicaps majeurs, mais n'affecte pas l'espérance de vie. - SMA type 4 : c'est la forme adulte de la maladie ainsi que la moins sévère ; il commence généralement vers la troisième décennie de la vie et a un cours très lent.
Elle n'est généralement pas responsable de problèmes respiratoires et est associée à une « espérance de vie normale ».
Les niveaux de protéine SMN affectent la sévérité de la SMA : plus la quantité de SMN est faible, plus la sévérité de la maladie associée est élevée.
La diminution des taux de SMN est étroitement liée à l'étendue du défaut génétique affectant les gènes SMN1 ou SMN2 : plus ce défaut est étendu, plus la diminution de la quantité de protéine SMN est importante (c'est le cas, par exemple, d'une délétion de gène).
De plus, l'AMS ne compromet pas les fonctions intellectuelles (le QI des patients est normal) et épargne l'organe de la vue.
Pour en savoir plus : SMA : tous les SymptômesSymptômes SMA de type 0
Comme indiqué précédemment, l'AMS de type 0 survient déjà à l'âge prénatal avec une mobilité fœtale réduite ; à la naissance, l'enfant malade présente donc des difficultés évidentes à avaler et à respirer.
La maladie entraîne la mort quelques semaines après la naissance, même lorsque le patient reçoit une assistance respiratoire.
Symptômes de l'AMS de type 1
Les enfants atteints d'AS de type 1 ont des muscles très faibles qui ne se développent pas comme ils le devraient (atrophie musculaire). Cela les empêche de faire des activités telles que lever la tête, bouger les membres et s'asseoir; de plus, il complique progressivement les fonctions vitales, telles que la succion du lait, la déglutition, la mastication et la respiration.
En règle générale, l'AMS de type 1 est fatale au cours des premières années de la vie ; certains patients parviennent cependant à atteindre l'âge de l'adolescence.
La mort survient généralement à la suite d'une insuffisance respiratoire ou d'une « infection pulmonaire due à des difficultés de déglutition (pneumonie d'ingestion ou pneumonie ab ingestis).
Symptômes de l'AMS de type 2
Le SMA de type 2 se manifeste classiquement par :
- Douceur des muscles des bras et des jambes;
- Tremblements dans les doigts et les mains;
- Difficulté à assumer la position assise de manière autonome (le patient parvient cependant à la maintenir);
- Difficulté à se tenir debout et à marcher
- Malformations et problèmes articulaires ;
- Difficulté à respirer et à avaler des aliments ;
- Scoliose (apparaît généralement plus tard).
Même dans cette situation, les difficultés respiratoires et la déglutition des aliments sont à l'origine de décès prématurés, qui surviennent généralement au début de l'âge adulte.
Symptômes de l'AMS de type 3
L'AMS de type 3 provoque des problèmes de posture et d'équilibre, des tremblements de la main et des difficultés à se lever d'une position assise, à marcher, à monter les escaliers et à courir.
Au début, les affections ne nécessitent pas d'aide à la locomotion ; par la suite, avec la dégénérescence d'un plus grand nombre de motoneurones, les béquilles, les déambulateurs et les fauteuils roulants deviennent fondamentaux.
Bien que cela puisse arriver, il est très rare que les patients atteints d'AS de type 3 souffrent de problèmes respiratoires et d'ingestion de nourriture.
En présence de cette forme de SMA, l'espérance de vie est normale, mais avec tous les problèmes susmentionnés.
Symptômes de l'AMS de type 4
À l'âge adulte, l'AMS de type 4 est généralement associée à :
- Affaiblissement du tonus musculaire dans les bras et les jambes;
- Difficulté à marcher
- Tremblements et secousses musculaires soudaines.
Initialement, les plaintes susmentionnées sont modérées ; dans la vieillesse, ils deviennent plus cohérents.
Comme l'AMS de type 3, l'AMS de type 4 n'est pas une maladie qui affecterait l'espérance de vie du patient.
SMA : quand consulter un médecin ?
Il est fortement conseillé à tous les parents qui se savent porteurs sains de SMA de consulter un pédiatre spécialisé dans les maladies génétiques et un généticien.
Si vous ne disposez pas d'informations de ce type, il est bon d'évaluer mois par mois le développement moteur de votre enfant et les fonctions dont dépend la vie (ex : respiration).
Certes, l'incapacité de s'asseoir ou d'assumer la position assise, la difficulté à s'alimenter, la présence de déficits respiratoires et une musculature fine et moins tonique que celle des pairs constituent des sonnettes d'alarme.
Comme pour la forme adulte de l'AMS, l'apparition plus ou moins brutale d'une faiblesse musculaire et d'une difficulté à marcher est suspectée et à surveiller.
Amyotrophie spinale : complications
Les formes les plus sévères de l'AMS peuvent entraîner des complications telles que :
- Suffocation due à la nourriture. Cela est dû à la capacité réduite à mâcher et à ingérer des aliments.
- Arrêt respiratoire. C'est une conséquence de l'incapacité de contrôler l'activité des muscles respiratoires.
- Pneumonie ab ingestis (ou pneumonie par inhalation). Elle survient lorsqu'un corps étranger porteur d'agents pathogènes, comme de la nourriture, de la salive ou des sécrétions nasales, pénètre ou s'accumule dans les poumons.
Pneumonie ab ingestis c'est le résultat de difficultés de déglutition. - Paralysie entraînant l'utilisation de fauteuils roulants. Elle survient lorsque la maladie a irrémédiablement compromis les facultés locomotrices du patient.
- Malnutrition. C'est une autre conséquence de la difficulté à avaler : le patient, en effet, peine à s'alimenter correctement.
Il convient de noter que, parfois, des tests tels que l'électromyographie ou la biopsie musculaire peuvent être utilisés lors du diagnostic de l'AMS.
SMA : Examen physique et anamnèse
L'examen physique chez un patient susceptible de souffrir d'AMS implique une analyse minutieuse des symptômes et la recherche de certains signes typiques de la maladie, tels que :
- Faiblesse et sensibilité des muscles ;
- Contractions musculaires soudaines
- Réflexes tendineux réduits ou absents.
En ce qui concerne les antécédents médicaux, cependant, cela se concentre principalement sur les antécédents familiaux du patient, afin d'établir si un autre membre de la famille (parents, frères et sœurs, grands-parents) se plaint ou se plaint d'une symptomatologie similaire. maladie, transmise par les parents.
Bien qu'ils ne permettent pas d'établir un diagnostic définitif, l'examen physique et les antécédents médicaux peuvent fournir des informations très utiles, qui orientent les investigations vers la réalisation d'un test génétique.
De toute évidence, si le patient est un petit enfant, les parents interagiront avec le médecin au cours de l'histoire médicale.
SMA et test génétique

Le test génétique pour la détection de la SMA implique la recherche et l'étude des mutations des gènes SMN1/SMN2 dans un échantillon de cellules sanguines du patient.
La présence d'altérations génétiques signifie évidemment la maladie.
L'analyse des mutations détectées est essentielle pour établir le type d'amyotrophie spinale présente et la gravité de l'affection.
Pour connaître les résultats du test génétique précité, il faut généralement attendre de 3 à 4 semaines (les délais d'attente précis varient selon le centre génétique qui réalise le test).
SMA : le diagnostic prénatal est-il possible ?
Il est possible de diagnostiquer l'AMS à l'âge prénatal.
Pour ce faire, vous avez besoin d'un test génétique sur un échantillon de cellules fœtales, obtenu par des méthodes délicates telles que la villocentèse ou l'amniocentèse.
Compte tenu du risque d'avortement qui caractérise le CVS et l'amniocentèse, les médecins poursuivent la recherche prénatale de toute mutation attribuable à « l'amyotrophie spinale uniquement s'il existe des antécédents familiaux de SMA ou si l'enfant à naître est l'enfant de porteurs sains de la maladie.
SMA et dépistage néonatal
Il convient de noter que dans quelques régions italiennes (Lazio et Toscane) un service est actif dépistage pour le diagnostic précoce de la SMA et d'autres maladies génétiques graves.
Le diagnostic précoce de ces maladies permet de planifier en temps opportun le traitement symptomatique le plus approprié pour le contrôle des symptômes et des complications.
Amyotrophie spinale et planification d'une grossesse
Le conseil génétique est recommandé pour toutes les femmes en quête de grossesse qui :
- Ils ont eu un enfant atteint de SMA lors d'une grossesse précédente ;
- Ils ont des antécédents familiaux de SMA derrière eux ;
- Sont-ils porteurs sains de la maladie ou leur partenaire l'est.
Le conseil génétique peut aider les femmes atteintes de ces maladies à comprendre les risques auxquels un futur enfant est exposé.
SMA et diagnostic différentiel
Il existe deux pathologies très proches de l'AMS, que seule une "investigation diagnostique approfondie reconnaît et évite la confusion avec" l'amyotrophie spinale : il s'agit de "l'amyotrophie spinale avec détresse respiratoire (SMARD) et" l'amyotrophie bulbo-spinale (BSMA). maladie de Kennedy); la première est due à une mutation du gène IGHMBP2 situé sur le chromosome 11, tandis que la seconde est due à une mutation du chromosome sexuel X.
et produits pharmaceutiques) a approuvé Zolgensma, la méthode de thérapie génique pour le traitement de l'amyotrophie spinale.
Zolgensma consiste en une technique de biologie moléculaire très avancée, qui comprend l'utilisation d'un virus-vecteur capable d'insérer une copie normale du gène SMN1/SMN2 dans l'ADN présent à l'intérieur des motoneurones d'un patient.
L'administration du virus-vecteur précité s'effectue par injection intraveineuse.
Zolgensma s'est avéré efficace. Cependant, comme prévu, il présente deux limites principales qui empêchent son utilisation courante :
- C'est très cher. On parle de millions d'euros ;
- Il ne s'applique qu'aux patients SMA de moins de 2 ans.
Amyotrophie spinale : traitements symptomatiques
Les thérapies symptomatiques de l'AMS garantissent de plus grands bénéfices si elles sont adoptées rapidement ; cela rend le diagnostic précoce de la maladie très important.
SMA et assistance respiratoire
Une assistance respiratoire appropriée aide non seulement les personnes souffrant de SMA à respirer, mais réduit également le risque d'infections pulmonaires.
Parmi les différentes options thérapeutiques, on retrouve les masques pour la ventilation non invasive et les solutions plus invasives comme l'intubation orotrachéale et la trachéotomie ; les premières sont idéales pour les cas moins sévères, tandis que les solutions plus invasives sont indispensables pour les patients présentant de graves problèmes respiratoires.
SMA et soutien nutritionnel
Les formes les plus sévères d'amyotrophie spinale affectent la capacité d'avaler et de mâcher des aliments, exposant le patient au risque d'étouffement, de pneumonie par ingestion et de malnutrition.
Pour contrôler ces conséquences dangereuses, il est essentiel de recourir à des aides à l'alimentation, comme une sonde nasogastrique ou une chirurgie de gastrostomie, et de s'appuyer sur un nutritionniste qui planifiera une alimentation adaptée aux besoins du patient.
SMA et physiothérapie
Les difficultés motrices qui caractérisent le patient atteint d'amyotrophie spinale entraînent une raideur articulaire et musculaire due à l'inactivité.
Un programme de physiothérapie adéquat permet d'améliorer, dans la mesure du possible, la souplesse des muscles et de rendre les articulations moins rigides.
Il est clair que ce programme comprend des exercices dont l'exécution est à la portée des capacités du patient.
SMA et orthopédie
En présence de scoliose, typique des formes sévères d'AMS, il est indispensable de consulter un orthopédiste ; ce dernier pourrait indiquer l'utilisation d'un corset orthopédique, si la déformation est légère, ou opter pour la chirurgie de fusion vertébrale, si la malformation vertébrale est sévère.
Médicaments contre la SMA
Depuis quelques années, il existe également des médicaments spécifiques contre l'AMS.
Ces médicaments méritent un traitement à part par rapport aux thérapies symptomatiques, bien qu'ils ne permettent pas de guérir la maladie, mais seulement de la contenir.
Les médicaments spécifiques contre la SMA actuellement disponibles sont le Spinraza (nusinersen) et l'Evrysdi (risdiplam) : le premier agit en corrigeant la production aberrante de la protéine SMN dans le processus ; le second augmente les niveaux de production de SMN, essayant également de les maintenir à un niveau quota adéquat aux besoins de l'organisme humain.
Approuvés par la FDA respectivement en 2017 et 2020, Spinraza et Evrysdi garantissent des résultats, dans certains cas même plus que satisfaisants, cependant ils ont une limitation importante : ils sont très coûteux.
Pour plus d'informations : Spinraza : Comment ça marche, risques et avantages