Généralité
Le syndrome de Crouzon est une maladie génétique rare, qui détermine la présence d'une craniosténose et d'autres anomalies faciales assez particulières.
Son apparition est causée par certaines altérations de l'ADN qui constituent les gènes FGFR2 et FGFR3 ; ces éléments génétiques sont impliqués dans le processus de maturation osseuse au cours du développement embryonnaire.

La thérapie consiste en une série d'interventions chirurgicales visant à résoudre les symptômes les plus importants et les plus dangereux.
Actuellement, le pronostic tend à être très souvent positif.
Examen de la génétique
Avant de procéder à la description du syndrome de Crouzon, il est utile de passer en revue quelques concepts fondamentaux de la génétique.
Qu'est-ce que l'ADN ? C'est le patrimoine génétique, dans lequel sont inscrits les traits somatiques, les prédispositions, les qualités physiques, le caractère, etc. d'un organisme vivant. Il est contenu dans toutes les cellules du corps ayant un noyau, comme se trouve à l'intérieur de cela.
Que sont les chromosomes ? Selon la définition, les chromosomes sont les unités structurelles dans lesquelles l'ADN est organisé. Les cellules humaines contiennent, dans leur noyau, 23 paires de chromosomes homologues (22 de type autosomique non sexuel et une paire de type sexuel) ; chaque paire est différente d'une autre, car elle contient une séquence génétique spécifique.
Que sont les gènes ? Ce sont de courts segments, ou séquences, d'ADN ayant une signification biologique fondamentale : d'eux, en fait, dérivent des protéines ou des molécules biologiques fondamentales pour la vie. Dans les gènes, il y a une partie « écrite » de qui nous sommes et de qui nous deviendrons.
Chaque gène est présent sous deux versions, les allèles : un allèle est d'origine maternelle, donc transmis par la mère ; l'autre allèle est d'origine paternelle, donc transmis par le père.
Qu'est-ce qu'une mutation génétique C'est une erreur dans la séquence d'ADN, qui forme un gène. Du fait de cette erreur, la protéine résultante est soit défectueuse, soit totalement absente.Dans les deux cas, les effets peuvent être délétères à la fois pour la vie de la cellule, dans laquelle se produit la mutation, et pour celle de l'organisme dans son ensemble. Les maladies congénitales et les néoplasmes (c'est-à-dire les tumeurs) appartiennent à une ou plusieurs mutations génétiques.
Le syndrome de Crouzon aussi
Le syndrome de Crouzon est une maladie génétique rare caractérisée par une craniosténose et le « développement non naturel de certains éléments du visage, notamment les yeux, le nez, la mâchoire et la mâchoire ».
C'est une maladie congénitale, dont les caractéristiques typiques peuvent être évidentes dès les premiers instants de la vie.
SIGNIFICATION DE CRANIOSINOSTOSI
La craniosynostose est le terme par lequel les médecins désignent la fusion prématurée d'une ou plusieurs sutures crâniennes.

Depuis le site : thecraniofacialcenter.com
Les sutures crâniennes sont les articulations fibreuses qui relient les os de la voûte crânienne (c'est-à-dire les os frontaux, temporaux, pariétaux et occipitaux).
Dans des conditions normales, la fusion des sutures crâniennes se produit dans la période postnatale (certains processus se terminent même à l'âge de 20 ans). Ce long processus de fusion permet au cerveau de grandir et de se développer adéquatement.
Si, comme dans le cas des craniosynostoses, la fusion a lieu trop tôt - donc au cours de la vie prénatale, périnatale* ou de la petite enfance - les éléments cérébraux (cerveau, cervelet et tronc cérébral) et certains organes des sens (yeux notamment) subissent une « altération de forme et de croissance.
* Le terme périnatal désigne la période de la vie qui va de la 27e semaine de gestation aux 28 premiers jours après l'accouchement.
ORIGINE DU NOM
Le syndrome de Crouzon doit son nom au médecin français Octave Crouzon, qui a le mérite d'en avoir décrit le premier ses principales caractéristiques cliniques.
Crouzon a vécu entre la fin des années 1800 et le début des années 1900, exactement de 1874 à 1938. Initialement, pour définir le syndrome qui a plus tard pris son nom, il a utilisé le terme de dysostose craniofaciale.
Causes
Le syndrome de Crouzon résulte d'une mutation du gène FGFR2, situé sur le chromosome 10, ou du gène FGFR3, situé sur le chromosome 4.
FGFR est l'acronyme anglais de Récepteur du facteur de croissance des fibroblastes, qui se traduit en italien est : Receptor for the Fibroblast Growth Factor.
Le rôle fonctionnel des gènes FGFR2 et FGFR3 est de produire chacun une protéine réceptrice, qui à son tour a pour tâche de réguler la maturation et le développement embryonnaire du tissu osseux.
Selon les théories des chercheurs, les mutations dans FGFR2 et FGFR3 hyper-stimuleraient ces mêmes gènes, qui, une fois de plus actifs, induiraient une maturation précoce de certains tissus osseux, dont ceux constituant le crâne.
LA GÉNÉTIQUE
Les mutations génétiques responsables du syndrome de Crouzon peuvent être héréditaires ou peuvent survenir spontanément après la conception.
Dans le premier cas, l'état morbide - que les médecins appellent également syndrome de Crouzon héréditaire - présente toutes les caractéristiques d'une maladie génétique autosomique dominante (ou maladie héréditaire dominante). Pour un lecteur novice de génétique, cela signifie que :
- La maladie et ses symptômes surviennent également en présence d'un seul allèle du gène muté (peu importe qu'il provienne de la mère ou du père), car ce dernier est dominant sur le sain.
- Un parent porteur de la mutation suffit pour avoir la maladie dans une partie de la descendance.
- La probabilité qu'un enfant malade naisse, issu d'un couple où un seul des deux composants porte la mutation, est de 50 %.
Dans le second cas, cependant, l'état morbide - que les experts indiquent avec la terminologie du syndrome de Crouzon non héréditaire - est le résultat d'un événement sporadique anormal, qui altère l'ADN au cours de la croissance embryonnaire du fœtus.
Résumé de la signification des termes héréditaire, autosomique et dominante
- Héréditaire : cela signifie que les parents transmettent l'altération génétique responsable de la maladie à la descendance (c'est-à-dire aux enfants).
- Autosomique : cela signifie que la mutation responsable de la maladie réside dans un chromosome non sexuel, donc autosomique.
- Dominant : signifie que la maladie provoque des symptômes et des signes même lorsqu'un seul allèle du gène responsable est muté. En termes plus simples, c'est comme si l'allèle avec la mutation avait plus de puissance que l'allèle sain.
ÉPIDÉMIOLOGIE
Selon certaines estimations du taux d'incidence du syndrome de Crouzon, environ un enfant sur 60 000 naîtrait avec cette maladie rare.
Le syndrome de Crouzon représente 4,5% des cas de craniosténoses.
Symptômes et complications
Les patients atteints du syndrome de Crouzon ont un tableau symptomatique très spécifique, qui consiste généralement en :
- Problèmes liés à la craniosténose, notamment :
-

De https://en.wikipedia.org/wiki/Plagiocephaly Brachycéphalie, qui est la compression de l'arrière de la tête. La fusion prématurée des sutures crâniennes coronales s'ensuit (craniosynostose coronale).
Si elle n'est pas traitée, elle peut affecter la croissance du cerveau et le développement des capacités cognitives.
Elles représentent une « alternative à la brachycéphalie : la trigonocéphalie (fusion de la suture métopique), la dolichocéphalie (fusion de la suture sagittale) et la plagiocéphalie (fusion des sutures coronales). - Exophtalmie, qui est le terme pour la saillie des globes oculaires. Cela pourrait impliquer la présence de problèmes de vision.
- Hypertélorisme oculaire, c'est-à-dire des yeux exagérément éloignés les uns des autres. Avec l'exophtalmie, cela peut aggraver les problèmes de vision.
- Nez déformé, généralement en forme de bec. Si elle est grave ou non traitée chirurgicalement, cette anomalie peut entraîner des problèmes respiratoires ou les mêmes symptômes que le syndrome d'apnée obstructive du sommeil.
- Augmentation de la pression intracrânienne. Elle est également connue sous le nom d'hypertension intracrânienne. Sa présence s'explique par le fait que les structures cérébrales n'ont pas le bon espace pour se développer.
Habituellement trouvée au milieu de l'enfance, l'hypertension intracrânienne est une cause potentielle de maux de tête, de vomissements et de douleurs oculaires. - Hydrocéphalie, qui est le résultat d'une augmentation du liquide céphalo-rachidien contenu dans l'espace sous-arachnoïdien et dans les ventricules cérébraux.
- Malformation d'Arnold-Chiari (ou syndrome d'Arnold-Chiari). C'est une déformation située à la base du crâne.
* L'hydrocéphalie et la malformation d'Arnold-Chiari sont généralement deux complications qui surviennent en l'absence de traitements adéquats. - Anomalies de la mandibule et du maxillaire.
Le premier a des dimensions plus petites que la normale, tandis que le second a tendance à faire saillie vers l'extérieur.Tout cela modifie la forme du palais et de l'échafaudage dentaire (absence de certaines dents, etc.), avec des répercussions (parfois même graves) sur la phonation et sur la mastication.
Certains patients naissent avec une fente labiale (fente labiale) ou une fente palatine.
- Problèmes d'audition.
55% des patients atteints du syndrome de Crouzon naissent sans conduits auditifs ou avec des anomalies majeures. Il en résulte une capacité acoustique absente ou fortement réduite.
Certains sujets développent un ensemble de problèmes auditifs à l'âge adulte, attribuables au tableau clinique typique du syndrome de Ménière.
- Problèmes articulaires dans le cou.
Ils concernent 30% des cas de syndrome de Crouzon.
- Anomalies de la peau.
Les patients atteints d'un syndrome de Crouzon muté soutenu par le FGFR3 présentent acanthosis nigricans, une dermatose caractérisée par une augmentation de l'épaisseur (hyperkératose) et un assombrissement (hyperpigmentation) de la peau.

Deux autres anomalies anatomiques associées (quoique rarement) au syndrome de Crouzon
- Canal artériel breveté
- Coarctation de l'aorte
SYNDROME DE CROUZON ET QI
Grâce également aux possibilités actuelles de traitement de la craniosténose, 97 % des patients atteints du syndrome de Crouzon ont aujourd'hui une « intelligence normale ».
Diagnostic
Un pédiatre expérimenté peut être en mesure de diagnostiquer le syndrome de Crouzon au moyen de l'examen physique seul.
En présence de tout doute ou perplexité, les éléments suivants sont fondamentaux pour arriver à une conclusion précise :
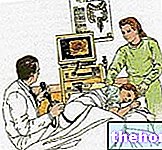
- Images radiologiques, fournies par des radiographies ou des tomodensitogrammes de la tête
- Un test génétique, visant à rechercher d'éventuelles mutations de l'ADN.
EXAMEN OBJECTIF
L'examen physique consiste en une analyse précise de la tête et des anomalies présentes sur celle-ci.
Les déformations crâniennes, induites par une craniosténose (par exemple brachycéphalie), sont parmi les signes cliniques les plus caractéristiques du syndrome de Crouzon et sur lesquelles le médecin fonde une partie de ses conclusions diagnostiques.
EXAMENS RADIOLOGIQUES
Les radiographies et les tomodensitogrammes de la tête montrent quelles sutures crâniennes ont fusionné prématurément.
La craniosténose qui caractérise le syndrome de Crouzon affecte les sutures coronales, donc une fusion retrouvée au niveau de ces dernières est une information très souvent déterminante pour le diagnostic.
EXAMEN GÉNÉTIQUE
En plus de montrer si l'ADN a des mutations, les tests génétiques permettent d'identifier le gène exact qui cause le syndrome de Crouzon, qu'il s'agisse du FGFR2 ou du FGFR3.
Traitement
Aujourd'hui, les porteurs du syndrome de Crouzon peuvent compter sur divers traitements, en fonction de la gravité de l'affection et des symptômes.
En effet, les médecins ont assuré :
- Chirurgie pour la résolution de la craniosténose et de ses symptômes.
- Aides acoustiques, en cas de problèmes auditifs.
- Thérapies pour l'amélioration des compétences linguistiques.
- Thérapies chirurgicales pour l'amélioration des anomalies du maxillaire et de la mandibule.
- Une chirurgie, connue sous le nom de trachéotomie, pour résoudre les problèmes respiratoires.
Veuillez noter: Le syndrome de Crouzon est une affection morbide qui découle d'une « altération génétique de l'ADN impossible à guérir. Ainsi, en fait, les médecins ne traitent la maladie que d'un point de vue symptomatologique.
CHIRURGIE DE LA CRANIOSYNOSTOSE
Les objectifs thérapeutiques de l'intervention chirurgicale sont au nombre de deux :
- Donnez aux structures cérébrales et aux yeux l'espace dont ils ont besoin pour se développer et fonctionner au mieux.
- Donnez à la tête une forme normale, puis résolvez le problème de la brachycéphalie.
Les chirurgiens ont la possibilité de réaliser l'opération de deux manières (ou approches) différentes : par une "opération de chirurgie traditionnelle - également appelée" à ciel ouvert "- ou par une "opération de chirurgie endoscopique".
La « chirurgie ouverte » consiste en la « réalisation d'une » incision sur la tête, par laquelle le médecin opérateur extrait l'os ou les os crâniens malformés qui doivent être remodelés. A la fin du remodelage, le chirurgien réinsère les structures osseuses préalablement extraites et referme l'incision par des sutures.
La chirurgie endoscopique, quant à elle, implique l'utilisation d'un endoscope et la pratique d'une très petite incision sur la tête, à travers laquelle le médecin opérateur insère lui-même l'endoscope.
L'endoscope est en fait un tube fin et flexible, équipé d'une caméra à fibre optique (à l'extrémité insérée dans le crâne) et relié à un moniteur. Grâce à cet instrument particulier et aux images qu'il projette sur le moniteur, le chirurgien est en mesure de séparer prématurément les sutures crâniennes de fusion, avec une précision remarquable et sans recourir à des incisions cutanées et des extractions osseuses.
Selon les experts, le meilleur moment pour effectuer la chirurgie est pendant la toute petite enfance (les 12 premiers mois de la vie), car les os se moulent plus facilement.
Cependant, il faut rappeler que plus le patient est jeune, plus le risque de récidive des mêmes sutures crâniennes (récidive) est élevé.En cas de récidive, la chirurgie doit être répétée.
Selon certaines recherches statistiques, 10 à 20 % des sujets très jeunes, subissant une chirurgie de la craniosténose, doivent subir une deuxième opération, en raison d'une rechute.
TRAITEMENT DES PROBLÈMES ACOUSTIQUES
En plus de prescrire l'utilisation d'appareils auditifs, les médecins recommandent également des contrôles auditifs périodiques, car c'est le meilleur moyen de prévenir toute aggravation des problèmes existants.
THÉRAPIES CHIRURGICALES POUR LA MÂCHOIRE ET LES ANOMALIES DE LA MÂCHOIRE
Le traitement des anomalies maxillaires et mandibulaires comprend la chirurgie pour le réalignement du maxillaire et/ou de la mandibule, certains traitements dentaires pour la disposition des arcades dentaires et l'opération pour la résolution de la fente labiale et/ou de la fente palatine.
TRACHÉOTOMIE
La trachéotomie est l'opération chirurgicale par laquelle le médecin crée, au niveau du cou (où passe la trachée), un passage pour l'air destiné aux poumons. Cela permet à ceux qui subissent cette chirurgie de respirer à nouveau et correctement.
Pour transporter l'air dans les poumons, vous avez besoin d'un petit tube, appelé tube de transchéotomie, qui est de la bonne taille pour être inséré dans la trachée.
Pronostic
En général, le pronostic dépend de la sévérité de la craniosténose : si cette dernière est traitable avec de bons résultats, les patients atteints du syndrome de Crouzon peuvent mener une vie presque normale.



-cos-cause-e-terapia.jpg)