Définition
La maladie de Wilson - également connue sous le nom de dégénérescence hépatolenticulaire - est une maladie génétique rare et héréditaire. Cette maladie provoque une accumulation de cuivre dans les tissus et les organes des individus atteints.
Les effets les plus importants de cette accumulation anormale se manifestent surtout dans le cerveau et le foie, mais pas seulement.
Si elle n'est pas correctement traitée, la maladie de Wilson peut entraîner des issues fatales.
Causes
S'agissant d'une maladie génétique, la cause déclenchante réside dans l'altération d'un gène.Plus précisément, il s'agit d'une modification du gène ATP7B situé sur le chromosome 13.
Ce gène code pour une protéine particulière, dont le rôle est de favoriser l'excrétion du cuivre en excès par la bile.Du fait de cette modification génétique, la protéine en question n'est pas produite et, par conséquent, le cuivre s'accumule dans les organes et les tissus.
Symptômes
Une hépatite, une cirrhose du foie, un ictère, une hépatomégalie et une splénomégalie peuvent survenir chez les patients atteints de la maladie de Wilson. De plus, des symptômes tels que vomissements, douleurs abdominales, dysarthrie, dysphagie, tremblements, ralentissement des mouvements et difficulté à marcher, migraine, faiblesse et raideur musculaire, changements d'humeur et de personnalité, difficultés de concentration, dépression, hématurie, glycosurie, peuvent survenir. anémie, aménorrhée. et anneaux Kayser-Fleischer.
Les informations sur la maladie de Wilson - Médicaments et soins ne sont pas destinées à remplacer la relation directe entre un professionnel de la santé et un patient. Consultez toujours votre médecin et/ou votre spécialiste avant de prendre la maladie de Wilson - Médicaments et traitement.
Médicaments
Le traitement pharmacologique de la maladie de Wilson repose sur l'administration de médicaments capables de chélater le cuivre ou de réduire son absorption, de manière à favoriser son élimination de l'organisme.
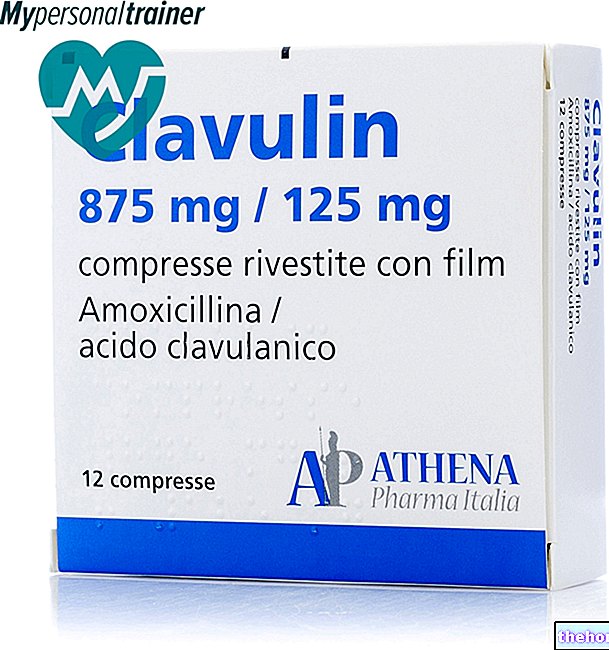
Le médicament de choix pour le traitement de la maladie de Wilson est la pénicillamine. Par ailleurs, dans certains pays européens (comme par exemple le Royaume-Uni), dans le cas où le traitement à la pénicillamine n'est pas toléré, une thérapie alternative à base de trientine (un autre médicament capable de chélater le cuivre, moins efficace mais avec moins d'effets secondaires que la pénicillamine).
Cependant, pour réduire l'absorption du cuivre, on administre généralement du zinc.
En plus du traitement médicamenteux, les patients atteints de la maladie de Wilson doivent également adopter un certain régime alimentaire, en veillant à éviter la consommation d'aliments riches en cuivre, tels que le chocolat, les noix, les champignons, le foie et les fruits de mer.
Dans le cas où la maladie de Wilson a causé des dommages irréparables au foie et/ou si le traitement médicamenteux n'est pas efficace, le médecin peut juger nécessaire de réaliser une greffe du foie.

Voici les médicaments les plus utilisés dans la thérapie contre la maladie de Wilson et quelques exemples de spécialités pharmacologiques ; il appartient au médecin de choisir le principe actif et la posologie les plus adaptés au patient, en fonction de la gravité de la maladie, de l'état de santé du patient et de sa réponse au traitement.
pénicillamine
La pénicillamine (Pemine®) est une molécule capable de chélater le cuivre et de favoriser son excrétion par l'urine. C'est un médicament disponible pour une administration orale avec des indications spécifiques pour le traitement de la maladie de Wilson. Sans surprise, c'est le médicament de premier choix qui est utilisé contre cette maladie génétique rare.
La dose de pénicillamine habituellement administrée chez l'adulte est de 15 à 40 mg/kg de poids corporel par jour, à prendre en quatre prises fractionnées une heure avant, ou 2-3 heures après les repas et en tout cas toujours à jeun.
Chez les enfants, en revanche, la dose de médicament habituellement utilisée est de 10 à 30 mg/kg de poids corporel, à prendre en 3 à 4 prises fractionnées de la même manière que pour les patients adultes.
Dans tous les cas, la posologie exacte du médicament doit être déterminée par le médecin pour chaque patient.
En outre, il convient de souligner que - compte tenu des effets secondaires que la pénicillamine peut provoquer - son utilisation doit toujours être sous la stricte surveillance du médecin.
Acétate de zinc
L'acétate de zinc (Wilzin ®) est un autre médicament ayant des indications thérapeutiques spécifiques pour le traitement de la maladie de Wilson.
Le zinc agit en réduisant l'absorption du cuivre dans l'intestin, en favorisant son élimination par les fèces et - par conséquent - en empêchant son accumulation dans le foie et d'autres organes et tissus.
L'acétate de zinc est disponible pour une administration orale sous forme de gélules.
La dose de médicament habituellement administrée aux patients adultes est de 50 mg, à prendre trois à cinq fois par jour au maximum, à jeun, une heure avant ou 2-3 heures après les repas.
Chez les enfants, cependant, la dose de médicament à administrer varie en fonction de l'âge et du poids corporel des patients.
Cependant, même dans ce cas, la posologie exacte du médicament doit être établie par le médecin au cas par cas.
Si un traitement à l'acétate de zinc est prescrit en même temps qu'un traitement à la pénicillamine, les deux médicaments doivent être administrés avec un intervalle d'au moins une heure entre l'un et l'autre.