La cause du syndrome d'Alport est la mutation de gènes impliqués dans la production d'une protéine essentielle au bon fonctionnement des reins, de l'oreille interne et des yeux.
Le diagnostic du syndrome d'Alport est basé sur un examen physique, des antécédents médicaux, une biopsie rénale et un test génétique.
Actuellement, les personnes atteintes du syndrome d'Alport ne peuvent compter que sur des traitements symptomatiques, c'est-à-dire qu'ils soulagent les symptômes et retardent les complications.
Le syndrome d'Alport est une maladie héréditaire, où le terme « hérité » signifie « transmis par l'un ou les deux parents ».
Épidémiologie : Quelle est la fréquence du syndrome d'Alport ?
Selon les statistiques, une personne sur 50 000 naît avec le syndrome d'Alport.
Synonymes du syndrome d'Alport
En raison de sa nature héréditaire et de son atteinte rénale, le syndrome d'Alport est également connu médicalement sous le nom de néphrite héréditaire.
les humains sont des séquences d'ADN qui ont pour tâche de produire des protéines fondamentales dans les processus biologiques essentiels à la vie, y compris la croissance et la réplication cellulaires.Lorsqu'ils sont exempts de mutations (donc chez une personne saine), les 3 gènes associés au syndrome d'Alport produisent un composant protéique fondamental pour la bonne génération, le traitement et la structuration finale du collagène de type IV qui se situe principalement dans les reins, dans l'oreille interne et dans les yeux.
En revanche, lorsqu'ils sont victimes de mutations, les 3 gènes liés au syndrome d'Alport perdent la capacité de générer le composant protéique indispensable à la réalisation dudit collagène de type IV et cela implique une « altération tissulaire au niveau des reins, de l'oreille interne et les yeux, avec des dysfonctionnements conséquents de la part de ces organes.
Saviez-vous que...
L'implication du collagène de type IV par le syndrome d'Alport fait de ce dernier une maladie du tissu conjonctif, comme le syndrome d'Ehlers Danlos ou le syndrome de Marfan.
Physiopathologie du syndrome d'Alport : quelques précisions supplémentaires
Les gènes liés au syndrome d'Alport contribuent à la formation d'une variante du collagène de type IV, qui est essentielle pour :
- La bonne action filtrante des glomérules rénaux vers le sang.
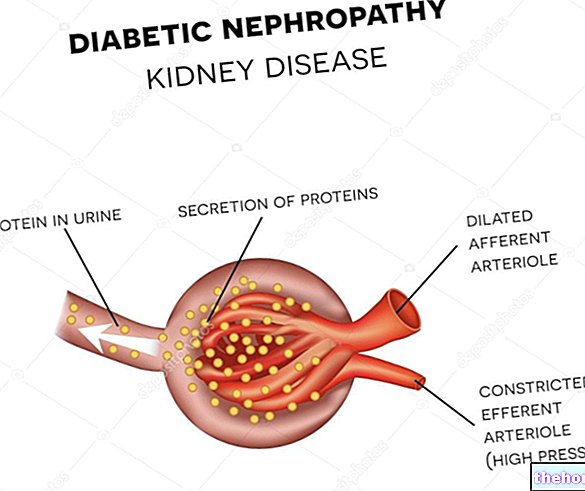
Explication. Situés dans les reins, les glomérules rénaux sont des agglomérations particulières de vaisseaux sanguins, capables d'éliminer (comme s'il s'agissait de filtres) les déchets du sang et de créer de l'urine.
Avec l'altération de leur collagène de type IV, ces structures rénales échouent dans leur action de filtrage et de création d'urine.

- Le travail de traduction des ondes sonores en impulsions nerveuses par l'organe Corti de l'oreille interne.
Explication. L'organe de Corti est la structure de l'oreille interne chargée de transformer les ondes sonores perçues par l'oreille en signaux nerveux « lisibles » et « interprétables » par le cerveau humain.
L'altération du collagène de type IV associée à l'organe de Corti affecte le processus de traduction du son dans le langage du système nerveux humain. - Le maintien de la forme correcte du cristallin, de la rigidité adéquate de la cornée et de la couleur normale de l'épithélium pigmenté de la rétine.
Explication. La forme du cristallin et la rigidité de la cornée sont essentielles pour la fonction visuelle et la santé oculaire (la couleur de l'épithélium pigmenté de la rétine, en revanche, ne semble pas avoir d'importance dans ces zones).
La présence d'un collagène de type IV altéré dans les yeux modifie la forme du cristallin, la rigidité cornéenne et la couleur de l'épithélium pigmentaire rétinien.
Toutes ces informations expliquent pourquoi le syndrome d'Alport est associé à une certaine symptomatologie.
Hérédité du syndrome d'Alport
Comprendre...
- Chaque gène humain est présent en deux copies, appelées allèles, l'une d'origine maternelle et l'autre d'origine paternelle.
- Une maladie héréditaire est autosomique dominante lorsqu'une seule copie du gène qui la provoque suffit à muter.
- Une maladie héréditaire est autosomique récessive lorsque les deux copies du gène qui la provoque doivent muter pour se produire.
Le syndrome d'Alport a 3 modèles différents d'hérédité.
Selon le modèle héréditaire le plus courant (80 % des cas), le syndrome d'Alport est une maladie dite héréditaire liée au chromosome X (telle que l'hémophilie ou le daltonisme), car elle dépend de la mutation du gène COL4A5 situé sur le chromosome sexuel X.
Selon le deuxième modèle de transmission le plus courant (environ 10 % des cas), le syndrome d'Alport se comporte comme une maladie héréditaire dite autosomique récessive, car sa présence nécessite une mutation spécifique dans les deux allèles de l'un des gènes COL4A3 et COL4A4, localisés sur le chromosome 2.
Enfin, selon le modèle héréditaire moins courant (environ 5 % des cas), le syndrome d'Alport représente un exemple de maladie héréditaire autosomique dominante, puisqu'une mutation spécifique dans un seul des allèles des gènes COL4A3 et COL4A4 suffit à son manifestation. .
Syndrome d'Alport lié à l'X
Le syndrome d'Alport lié à l'X a ses pires effets chez les hommes.
Ce phénomène est lié à la constitution chromosomique sexuelle qui caractérise les hommes et les femmes ; en effet, chez la femme, il existe deux chromosomes X, qui, en présence d'une mutation dans l'un des deux, s'entraident (le sain compense, plus ou moins efficacement, les déficiences du muté) ; chez l'homme, en revanche, il n'y a qu'un seul chromosome X (l'autre est un chromosome sexuel Y), qui, s'il est sujet à des mutations, ne peut compter sur le support d'aucun autre chromosome ayant les mêmes fonctions.
Syndrome d'Alport autosomique récessif
Le syndrome d'Alport avec comportement autosomique récessif provoque des symptômes et des signes similaires chez les hommes et les femmes.
Syndrome d'Alport autosomique dominant
Le syndrome d'Alport avec comportement autosomique dominant affecte les hommes et les femmes avec la même sévérité.
et de l'arbre trachéo-bronchique, et des problèmes vasculaires sévères (dissection aortique et anévrisme de l'aorte abdominale ou thoracique).Perte de la fonction rénale : symptômes
PrémissePar « perte progressive de la fonction rénale », les médecins entendent que les reins et leurs structures internes (par exemple les glomérules rénaux) sont soumis à une baisse progressive de leur capacité de filtration et de production d'urine.
En présence du syndrome d'Alport, les symptômes liés à la perte de la fonction rénale consistent principalement en une hématurie, c'est-à-dire du sang dans les urines, et une protéinurie, c'est-à-dire la présence de protéines dans les urines.
L'hématurie est un symptôme assez précoce, dans le sens où, chez les sujets atteints du syndrome d'Alport, elle a tendance à apparaître à un jeune âge ; dans la plupart des cas, sa reconnaissance n'est possible qu'au microscope (hématurie microscopique).
A l'inverse, la protéinurie est un symptôme tardif, c'est-à-dire qu'elle apparaît lorsque le syndrome d'Alport est à un stade avancé.
Baisse des capacités auditives : les détails

Au niveau auditif, le syndrome d'Alport provoque une surdité partielle ; pour être précis, il induit une « perte auditive neurosensorielle, qui empêche le patient d'entendre les sons à hautes fréquences (perte auditive haute fréquence).
L'évolution des troubles auditifs chez les personnes atteintes du syndrome d'Alport varie en fonction du gène sujet à mutation et du type de transmission ; en fait:
- Si la mutation réside dans COL4A5, les mâles développent une perte auditive vers la fin de la petite enfance, tandis que les femelles, dans les rares circonstances où elles sont affectées, la développent à un âge plus avancé ;
- Si la mutation réside dans COL4A4 ou COL4A3 et que la maladie est autosomique récessive, l'homme et la femme se plaignent des premiers symptômes de perte auditive à la fin de l'enfance ou au début de l'adolescence ;
- Si la mutation réside dans COL4A4 ou COL4A3 et que la maladie est autosomique dominante, les patients masculins et féminins développent une perte auditive neurosensorielle plus tard dans la vie.
Pour des raisons encore inconnues, le syndrome d'Alport épargne à certains patients des problèmes auditifs ; en d'autres termes, pour des raisons inconnues, certaines personnes atteintes du syndrome d'Alport ne développent aucune perte auditive neurosensorielle.
Anomalies oculaires et déficits visuels : les détails
Observables seulement chez un certain pourcentage de patients, les anomalies oculaires induites par le syndrome d'Alport sont à l'origine d'affections telles que : kératocône, lenticon, cataracte et la présence de taches sur la macula rétinienne.
A l'exception de la présence de taches sur la macula rétinienne, toutes ces affections qui viennent d'être évoquées affectent plus ou moins profondément les capacités visuelles des patients.
Léiomyomatose diffuse de l'œsophage et de l'arbre trachéo-bronchique
La léiomyomatose diffuse de l'œsophage et celle de l'arbre trachéo-bronchique sont des tumeurs bénignes rares qui, chez les sujets atteints du syndrome d'Alport, peuvent provoquer :
- Dysphagie ;
- Vomissements postprandiaux
- Douleur épigastrique et douleur rétrosternale ;
- Bronchite récurrente;
- Dyspnée;
- La toux;
- Cri perçant pendant la respiration.
Syndrome d'Alport : complications
La principale complication du syndrome d'Alport est l'état d'insuffisance rénale au dernier stade, résultant de la perte progressive et inexorable de la fonction rénale.
En médecine, le terme d'insuffisance rénale de « dernier stade » désigne le stade terminal ainsi que le stade le plus sévère de l'insuffisance rénale chronique, c'est-à-dire l'état dans lequel les reins ont perdu complètement et définitivement toute leur capacité fonctionnelle.
Responsable de conséquences très graves (ex : hypertension, œdème pulmonaire, fragilité osseuse, immunosuppression, atteinte du système nerveux, etc.), "l'insuffisance rénale de dernier stade nécessite un traitement chronique comme la dialyse et constitue l'indication la plus importante d'une" opération chirurgicale comme une greffe de rein.
Autres complications
D'autres complications possibles du syndrome d'Alport, qui ont cependant un taux de mortalité élevé, sont les hémorragies internes consécutives à la lacération d'une « éventuelle dissection aortique ou d'un » éventuel anévrisme de l'aorte thoracique ou abdominale.
rénal;Examen physique
C'est l'observation médicale des symptômes et des signes présentés par le patient.
Dans le cadre du syndrome d'Alport, il révèle une partie des problèmes rénaux, des problèmes auditifs (le cas échéant) et des anomalies oculaires (le cas échéant).
Anamnèse
C'est l'étude critique des symptômes à travers des questions spécifiques, combinée à un examen des antécédents familiaux du patient.
Dans le cadre du processus d'examen qui sert à reconnaître le syndrome d'Alport, l'anamnèse permet au médecin de connaître le moment exact de l'apparition des symptômes et de savoir si une maladie héréditaire survient dans la famille du patient.
Biopsie rénale
Elle consiste en la collecte et l'analyse en laboratoire d'un échantillon de cellules rénales.
Dans le cadre du syndrome d'Alport, il est utile d'évaluer l'état de santé des reins et de vérifier l'absence de collagène fonctionnel de type IV.
La biopsie rénale est une étape importante dans le diagnostic du syndrome d'Alport, car, si elle est opportune, elle permet de planifier le traitement le plus approprié dès que possible.
Test génétique
C'est l'analyse de l'ADN visant à détecter des mutations dans des gènes critiques.
Dans le contexte du syndrome d'Alport, il représente le test de diagnostic de confirmation et le test permettant d'établir le gène muté précis et le type de transmission.
Inhibiteurs de l'ECA
Normalement indiqués pour le traitement de l'hypertension et de certaines maladies cardiovasculaires, les inhibiteurs de l'ECA sont utilisés en présence du syndrome d'Alport, en raison de leur capacité démontrée à ralentir le déclin progressif des fonctions rénales, typique de la maladie héréditaire précitée.
Grâce à cette capacité, les inhibiteurs de l'ECA permettent donc de retarder l'apparition d'une insuffisance rénale ainsi que la nécessité de traitements importants tels que la dialyse ou la transplantation rénale.
Pour des raisons inconnues, les inhibiteurs de l'ECA sont inefficaces chez certaines personnes atteintes du syndrome d'Alport ; cela explique pourquoi les médecins recherchent des médicaments ayant un pouvoir similaire.
Thérapies pharmacologiques pour l'insuffisance rénale chronique
Lorsque le syndrome d'Alport a entraîné une insuffisance rénale chronique, il existe plusieurs médicaments utiles, notamment :
- Médicaments contre l'hypertension (parmi ceux-ci, les inhibiteurs de l'ECA et les ARA susmentionnés, et les diurétiques);
- Suppléments de calcium et de vitamine D (pour protéger les os des fractures);
- Sulfonate de polystyrène de sodium et analogues (pour empêcher l'accumulation de potassium dans le sang).
Dialyse et transplantation rénale
La dialyse est un traitement qui reproduit artificiellement certaines fonctions du rein, éliminant le sang des déchets et de l'eau en excès.
La transplantation rénale, quant à elle, est la chirurgie consistant à remplacer un ou les deux reins par un rein sain provenant d'un donneur compatible (le donneur peut être non vivant ou vivant).
La dialyse et la transplantation rénale sont toutes deux indiquées lorsque le syndrome d'Alport est à ses stades les plus avancés, c'est-à-dire lorsqu'il existe une insuffisance rénale au dernier stade.
Dispositif d'écoute pour malentendant
En présence du syndrome d'Alport, la prothèse auditive est un remède adapté à tous les patients atteints de surdité partielle.
Traitement du kératocône, du lenticone et de la cataracte
Lorsque le syndrome d'Alport affecte les yeux, provoquant kératocône, lenticon et/ou cataracte, les patients peuvent compter sur une chirurgie oculaire dont les résultats sont plus que satisfaisants.
Quelles figures médicales la Thérapie Symptomatique du Syndrome d'Alport implique-t-elle ?
Le traitement symptomatique du syndrome d'Alport nécessite l'intervention coordonnée de plusieurs médecins spécialistes, notamment : des pédiatres, des néphrologues, des audiologistes et des ophtalmologistes.




-cos-cause-e-terapia.jpg)